
Abstract
Background and Objective
Characterizing nicotine pharmacokinetics is challenging in the presence of background exposure. We performed a combined retrospective population pharmacokinetic analysis of 8 trials, including exposure to Tobacco Heating System and cigarettes (both inhaled), nicotine nasal spray and oral nicotine gum.
Method
Data from 4 single product use trials were used to develop a population pharmacokinetic model with Phoenix® NLME™ and to derive exposure parameters. Data from 4 separate ad libitum use studies were used for external validation. A total of 702 healthy adult smokers (54% males; 21–66 years of age; smoking ≥10 cigarettes/day; from US, Europe and Japan) were eligible for participation.
Results
Two-compartment linear disposition combined with zero-order absorption model was adequate to describe nicotine pharmacokinetics, and a mono-exponentially decreasing background component was utilized to account for nicotine carry-over effects. Apparent nicotine clearance was typically 0.407 L/min in males and 26% higher in females (68% inter-individual variability). Bioavailability was product-specific, decreased with increasing nicotine ISO yield, and increased with increasing body weight. Absorption duration was apparently prolonged with nicotine gum. The typical initial and terminal half-lives were 1.35 and 17 h, respectively. The presence of menthol did not impact the determinants of the area under the curve. The model adequately described the external validation data.
Conclusions
The population model was able to describe in different populations the nicotine pharmacokinetics after single product use and after 4 days of ad libitum use of Tobacco Heating System, cigarettes, and of different nicotine replacement therapies with various routes of administration.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
A two-compartment linear disposition combined with zero-order absorption model was adequate to describe nicotine pharmacokinetics, and a mono-exponentially decreasing background component was utilized to account for nicotine carry-over effects. |
The presence of menthol did not impact product-specific bioavailability but only the apparent central volume (inversely related to C max) and duration of absorption (directly related to t max). |
The typical initial and terminal half-lifes were 1.35 and 17 h, respectively. |
1 Introduction
Cigarette smoking causes serious chronic diseases, including lung cancer, chronic obstructive pulmonary disease, cardiovascular disease and stroke [1, 2]. To reduce the morbidity and mortality attributable to smoking, tobacco control measures have been implemented in many countries. While smoking rates have been successfully reduced, they remain relatively high, and the World Health Organization estimates that there will be over a billion smokers by 2025 [3].
To complement tobacco control, approaches to tobacco harm reduction have been introduced with the development of alternative nicotine delivery systems, including the Tobacco Heating System 2.2 (THS 2.2). By heating instead of burning tobacco, THS avoids or reduces the formation of many toxic combustion products. Clinical studies have shown favorable biological effects in smokers who switched from cigarette to different non-combusted products [4–6]. While this makes risk reduction at the individual level possible, a population health benefit additionally depends on smokers actually switching from cigarettes to THS. A key requisite of THS or any other alternative nicotine delivery system thus is that it satisfies smokers and in particular furnishes a nicotine uptake profile similar to cigarettes [7], making pharmacokinetic assessment essential.
Clinical trials comparing the bioavailability of nicotine with THS 2.2, either mentholated or not, with various tobacco products (regular or mentholated CC, nicotine nasal spray (NNS), or mentholated nicotine gum), have been conducted in healthy adult smokers, in various populations. Data from 8 trials allowed for characterizing nicotine pharmacokinetics and sources of variability in a retrospective population analysis.
While designing single use cross-over trials, the duration of smoking abstinence prior to first product use and the duration of the washout period assumed that plasma nicotine terminal half-life (t 1/2, z) was approximately 2 h [8, 9]. However, in our clinical trials pre-dose nicotine plasma concentrations were often measurable in both study periods [7], resulting in carry-over. The reasons underlying the discrepancy between the literature-based half-life on the one hand and our findings of pre-dose and carry-over levels on the other hand might include: (1) high assay sensitivity in our studies; (2) prior background exposure; (3) insufficient duration of abstinence or washout periods; or (4) the nicotine half-life being longer than reported in the literature.
The objectives of this analysis were to (1) develop a population pharmacokinetic model describing plasma concentration–time profiles of nicotine based on single product use study data, (2) assess sources of variability in nicotine pharmacokinetic parameters, (3) estimate plasma exposure to nicotine, distinguishing between exposure due to product use and background exposure, and (4) assess the predictive performance of the nicotine population pharmacokinetic model in data from ad libitum use studies.
2 Methods
2.1 Clinical Studies
The full dataset was split into a learning and a validation dataset. The learning dataset was used for model building, covariate analysis and internal model evaluation. It was composed of 4 randomized, controlled, two-period cross-over, single center open-label confinement clinical trials. Healthy adult smokers of either sex were enrolled when eligible [10–13]. The objective of the studies was to compare nicotine pharmacokinetics and pharmacodynamics following single THS (regular or mentholated) versus CC (regular or mentholated) use in 44 subjects per study, and single THS (regular or mentholated) versus either regular NNS (outside of Japan) or mentholated nicotine gum (in Japan) use in 18 subjects per study (Table 1). The washout (smoking abstinence) period prior to period 1 was approximately 33–38 h and 48 h before period 2. Sixteen blood pharmacokinetic samples were taken in each period prior to the single product use, nominally 2, 4, 6, 8, 10, 15, 30, 45, 60, 120, 240, 360, 450, 720 and 1440 min after the use of THS, CC, and NNS. Accounting for the expected longer time to maximum concentration (T max), the first blood samples following NRT gum were taken at 10, 20, 25, 30, 35, 40, 45, 60, 120, 180 min.
The validation dataset included 4 ad libitum use studies conducted according to a common randomized, controlled, open-label, three-arm parallel group, multi-center design. Their objective was to evaluate if the ad libitum use of THS for 5 consecutive days affected the levels of biomarkers of exposure of selected HPHCs, compared to smoking CC and smoking abstinence. After a 2-day baseline period (Day 1 and Day 0) of CC ad libitum use, the exposure period in confinement consisted of 5 days of ad libitum use of the assigned product (CC or THS) in the THS and CC arms. Subjects in the smoking abstinence arm were not included in the present analysis. One blood sample for nicotine pharmacokinetic assessment was collected daily from Day 0 to Day 4, then within 15 min prior to first product use on Day 5. Up to 8 additional pharmacokinetic samples were taken on Day 5 and 2 samples on Day 6. The actual product use history was recorded on Days 5 and 6. Pharmacokinetic data collected prior to Day 5 were not included in the dataset.
Blood samples for the determination of nicotine concentrations in plasma were analyzed using a validated LC–MS/MS assay method. The lower limit of quantification (LLOQ) was 0.2 ng/mL for the single use studies and 1 ng/mL for the ad libitum studies.
2.2 Dataset Composition and Data Handling
The analysis dataset included all subjects having used a product at least once and having at least one measurable nicotine plasma concentration. The actual sampling times were used in the analysis. The analysis dataset included baseline demographic variables, daily cigarette consumption at baseline, scores of the Fagerström test for nicotine dependence (FTND) questionnaire, and some product-related information (e.g., type of product, presence of menthol, and nicotine dose). For inhaled products, the nicotine dose was defined as the nominal nicotine ISO yield of 0.5 mg for THS and varied for CC from 0.1 to 1.5 mg, depending on the individual brand. The nicotine dose was 2 mg in the mentholated nicotine gum and 1 mg for the NNS. Creatinine clearance was derived using the Cockcroft-Gault formula [14].
If an actual sampling time was missing, the nominal sampling time was imputed. No imputation was made for missing nicotine concentrations. Missing covariate values were imputed by the last observation carried forward. Nicotine plasma concentrations below the LLOQ were omitted from the analysis.
2.3 Population Pharmacokinetic Analysis
2.3.1 Base Model
The overall analysis process is illustrated in Fig. 1. The analysis was conducted according to applicable regulatory guidance documents [15, 16], using Phoenix® NLME™ 1.3 and first-order conditional estimation with extended least squares estimation.

Population pharmacokinetic analysis workflow. After omitting measurements below the level of quantification (BLQ) and splitting the full dataset into learning and validation datasets, an exploratory data analysis (EDA) was conducted to guide data cleaning and base model development. The covariate model (COV1) was developed sequentially after defining a hierarchy between primary and secondary covariates, until a final model was obtained. Sensitivity analyses assessed the impact of outlier data on the base model. Model evaluation was performed on the base and final models, including goodness-of-fit (GOF) plots. The final model was submitted to external evaluation using the validation dataset, and visual predictive check (VPC). Eventually, individual exposure metrics were derived from the learning dataset
All products were included simultaneously in an integrated population pharmacokinetic model. The data from two consecutive product use periods were analyzed jointly on a continuous time scale. A preliminary run was performed to detect potential outliers by visual inspection of concentrations associated with absolute conditional weighted residuals (CWRES) greater than 4 [17].
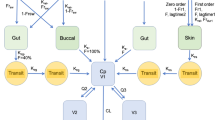
The base model for nicotine included two components sharing common disposition kinetics: a sub-model describing the time-course of background nicotine exposure, to address the previously observed carry-over effect, and a sub-model describing the time-course of plasma nicotine concentrations after product use. While neither of the components can be observed separately in the data, their combination (further referred to as “total concentration”) was used to model the observed plasma nicotine concentrations, as illustrated in Fig. 2.

Compartmental representation and equations defining the structural model of nicotine prior to inclusion of covariates. A1 nicotine amount in the central compartment, A2 nicotine amount in the peripheral compartment, Bckgrd model-predicted background nicotine concentration, alpha initial rate constant, beta terminal rate constant, C1 model-predicted concentration in the central compartment, C0 baseline nicotine concentration prior to first product use, C2 nicotine concentration in the peripheral compartment, Cl/F apparent clearance, Cl2/F apparent inter-compartmental clearance, Ctotal model-predicted total nicotine concentration, k10 microscopic elimination rate constant, k12 microscopic rate constant for the transfer from the central to the peripheral compartment, k21 microscopic rate constant for the transfer from the peripheral to the central compartment, Tdur duration of zero-order absorption, V1/F apparent central volume of distribution, V2/F apparent peripheral volume of distribution
The base model development considered either two- or three-compartment linear disposition, either first- or zero-order absorption kinetics, log-normally distributed pharmacokinetic parameters, either mixed (i.e., additional and proportional) or log-additive residual errors, and some combinations thereof. The background nicotine level was modelled by a decreasing mono-exponential term, assuming that product use occurred in the terminal phase of the preceding smoking abstinence or washout period. The model included inter-individual variability (IIV), as appropriate. Inter-occasion variability was, however, not considered as differences between periods were assigned to product-related differences, and captured by fixed-effect parameters.
Model evaluation and selection were based on a set of criteria including the log-likelihood (−2LL) difference between competing models, the Akaike Information Criterion (AIC), the precision of parameter estimation, successful model convergence, shrinkage of the empirical Bayes estimates (EBE) of the model parameters [18] and graphical goodness of fit representations.
2.3.2 Covariate Analysis
Whenever some a priori rationale for investigation was available, potential covariates were designated primary covariates, otherwise secondary covariates. Potential covariates, their hierarchy and the rationales for investigation are summarized in Supplementary Table 1 (Online Resource). The covariate analysis first addressed primary covariates, then extended to the secondary covariates to reduce the computational complexity [19].
The analysis of primary and secondary covariates followed a predefined scheme. First, a graphical exploratory data analysis (EDA) was conducted to detect trends. Then, based on the outcome, a subset of potential covariates was selected, considering biological plausibility and possible correlations between covariates, and ignoring clinically unimportant covariate effects in favor of model parsimony [20]. Finally, a forward selection/backward elimination procedure was used for the statistical covariate selection [21], with threshold p values of 0.05 and 0.01, respectively [19].
2.3.3 Model Evaluation and Sensitivity Analysis
An internal prediction-corrected visual predictive check (pcVPC) was performed based on the final population model, and observed concentrations in the learning dataset were compared to the corresponding simulated distributions [22]. The pcVPC was performed by period and product. For each stratum and time interval, percentile-based 90% prediction intervals (PIs) of the 10th, 50th and 90th percentiles of the simulated concentrations were computed across 1000 replicates and compared to the 10th, 50th and 90th percentiles of the observed concentrations.
The performance of the final population pharmacokinetic model to predict the external validation dataset was assessed using diagnostic plots [16, 18] after Bayesian post hoc estimation based on the actual product use history on Day 5/6.
A sensitivity analysis was performed to determine the potential impact of any outlier value excluded from the base model [15]. Parameter estimates obtained with the full population pharmacokinetic analysis dataset and the pharmacokinetic evaluation dataset were compared.
2.3.4 Individual Pharmacokinetic Parameters
To derive individual nicotine pharmacokinetic parameters, model-predicted concentration–time profiles (both total and background-adjusted) were simulated for each subject using empirical Bayes estimates (EBE). From these profiles, the following individual pharmacokinetic parameters were calculated by non-compartmental analysis using the Phoenix® WinNonlin® software, version 6.4: T max, initial half-life (t 1/2, 1), terminal half-life (t 1/2, z). In addition, both total and background-adjusted nicotine exposure pharmacokinetic parameters were derived: maximum concentration (C max), area under the concentration–time curve from time to 24 h (AUC0–24 h) and area under the concentration–time curve from time to infinity (AUCinf).
3 Results
3.1 Pharmacokinetic Population Analysis Set and Demographics
The pharmacokinetic population consisted of 728 subjects overall (Table 1), the analysis set including 702 subjects, i.e., 246 and 456 subjects in the learning and validation dataset, respectively, exclusions being due to discontinuation or lack of pharmacokinetic data or dosing history. Both datasets were comparable with regard to demographic characteristics, as summarized in Table 2.
3.2 Base Model
The learning dataset included 6843 measurable concentrations from 246 subjects, after exclusion of 831 values below the LLOQ and of 53 outliers (0.8%).
Two-compartment linear disposition was assumed based on the graphical data exploration and published models for orally [23] and transdermally or intravenously administered nicotine [24]. The prior knowledge of nicotine half-life, along with the length of the smoking abstinence and washout periods prior to product use warranted the selection of a mono-exponential sub-model for background nicotine despite multi-compartmental disposition. Macroscopic rate constants alpha and beta were derived from disposition parameters [25]. The background sub-model was defined by a baseline (C0) and a rate constant (beta). A three-compartment disposition did not improve the diagnostic plots.
Nicotine kinetics was better fit by zero- than by first-order absorption. The former involves a constant absorption rate over a finite duration (Tdur), consistent with the use of inhaled nicotine products and nicotine gum. Incidentally, zero-order absorption also adequately described nicotine absorption kinetics from the NNS. Considering that chewing would take longer than inhaling, the nicotine absorption duration (Tdur) was assumed to differ between nicotine gum and the other investigated products. In addition, the extent of absorption was assumed to differ between products. This was accounted for by including a product-specific bioavailability parameter (Frel), relative to THS as reference product (i.e., typical Frel set to 1).
The base model included IIV on all disposition parameters, as well as C0, Frel and Tdur. A log–additive residual error model was selected, facilitating parameter estimation and better accounting for the spread of observations, compared to competing error models.
Additional details related to the base model (development steps, model structure, equations, parameter estimates, and diagnostic plots) are provided in Fig. 2 and in Supplementary Tables 2 and 3 (Online Resource).
In a sensitivity analysis, parameters from the base model were re-estimated after introduction of the previously excluded 53 outlier concentrations. This model did not allow a successful covariance step and, given the minimal impact observed on parameter estimates (within ±14%), these observations were excluded from the remainder of the analysis.
3.3 Covariate Analysis
The covariate analysis was performed in the subset (6803 measurements in 244 subjects) of the learning dataset including only subjects with full covariate information (i.e., 2 subjects with missing CYP2A6 were excluded).
In the graphical EDA of primary covariates, the following effects were explored: effects of CYP2A6 activity and sex on both apparent clearance of apparent volume central compartment (Cl/F) and C0, and effects of body weight (WT) and nicotine ISO yield on Frel. All were found to be significant in the forward selection step (as assessed by the magnitude of the −2LL and AIC drops). The effect of sex on C0 was not retained in the backward elimination step.
In the subsequent graphical EDA of secondary covariates, the following effects were explored: age on apparent volume central compartment (V1/F), menthol on V1/F and Frel, race on C0 (being black versus non-black) and Tdur (being white versus non-white), and body height (HT) on Tdur. A residual effect of weight on C0 was also investigated. Only 3 of the above effects resulted in a significant drop in −2LL and were retained: The effects of menthol on V1/F and Frel, and of race on C0.
3.4 Final Model
The final population pharmacokinetic model was a two-compartment linear disposition model with zero-order absorption for product use and a mono-exponentially decreasing background component. For a typical subject from the learning dataset (male, not black, weight of 69.1 kg, using the regular variant of THS, baseline CYP2A6 activity of 29.2%) fixed effect estimates of V1/F, Cl/F, apparent volume peripheral compartment (V2/F), Cl2/F, Tdur and C0 were 70.0 L, 0.407 L/min, 171 L, 0.171 L/min, 5.30 min, and 0.358 ng/mL, respectively.
Compared to the base population pharmacokinetic model, the final model showed some improvement in IIV for Cl/F (68 versus 71%), V2/F (85 versus 100%), C0 (48 versus 57%), and for the effects of CC, NNS and nicotine gum on Frel (70 versus 79%), whereas IIV increased on Cl2/F (139 versus 135%). All fixed-effect parameters were precisely estimated with a low relative standard error (<4%) and shrinkage within 30%. The typical initial and terminal half-lives were 1.35 and 17 h, respectively (Table 3).
The final population pharmacokinetic model included effects of nicotine ISO yield, body weight and product type on nicotine bioavailability, CYP2A6 and sex on Cl/F, menthol on V1/F, baseline CYP2A6 activity and being black on baseline nicotine concentration, and menthol and nicotine gum on Tdur (Table 4).
The final population pharmacokinetic model did not include any effect of age, ALT, AST, height, body mass index, creatinine clearance, daily CC use, region (i.e., Japanese versus non-Japanese), total bilirubin, FTND total score and time to first cigarette (FTND item 1).
3.5 Model Evaluation
3.5.1 Internal Evaluation
Goodness-of-fit (GOF) plots and individual fits showed good agreement between observed and predicted nicotine concentrations, with no apparent bias (Supplementary Figs. 2 and 3, Online Resource).
PcVPC performed by product use and study period revealed that the model adequately captured the median pharmacokinetic profile, and the observed 90th percentile was generally within the prediction interval (PI), suggesting accurate model fit across products and time courses in both periods (Fig. 3). The model performance was harder to evaluate in the lower nicotine concentration range. Specifically, the 10th percentile was inconsistently captured by the simulations, possibly due to observations below the level of quantification (BLQ) being censored, whereas simulated concentrations were not.

Visual predictive check of the Final Model–Semi-log Scale (learning dataset). CC conventional cigarette, NNS nicotine nasal spray, PI prediction interval, THS tobacco heating system. Given the small number of measurements at 24 h, simulations were not displayed beyond 12 h for NNS and gum
3.5.2 External Model Evaluation
Out-of-(learning) sample evaluation of the final model was conducted in 456 validation dataset subjects, providing 4724 measurements after exclusion of 182 values BLQ (i.e., 3.7% of total measurements). Owing to a shorter smoking abstinence period in the ad libitum use studies of the validation dataset (typically 7.5 h overnight) compared to the single product use studies of the learning dataset (at least 31 h), the distribution of C0 was re-estimated in the validation dataset at 2.10 ng/mL (RSE = 2.0%). All other final population pharmacokinetic model estimates were fixed to final, and Bayesian post hoc estimation was performed. The resulting GOF plots appeared adequate (Supplementary Fig. 4, Online Resource).
3.6 Pharmacokinetic Parameters of Nicotine
The summary of exposure metrics derived from the simulated profiles by product type based on the final model is presented for the total and background-adjusted concentrations in Supplementary Table 4 (Online Resource). Across the learning dataset, background-adjusted exposure to nicotine was consistently lower with THS than with CC, on average by 24 or 26%, depending on whether based on C max or AUC, respectively.
4 Discussion and Conclusion
While nicotine metabolism and pharmacokinetics have been extensively studied [8, 9, 26–28], publications on nicotine pharmacokinetic models are limited. They include mechanistic or physiologically based pharmacokinetic models for nicotine, mainly to estimate nicotine dosimetry in different tissues such as the brain. Results were based on oral and/or intravenous nicotine administration in humans [23, 29, 30] or in rodents [31, 32], limiting the use of such data in the context of inhalation products.
Nicotine pharmacokinetics were adequately described by a two-compartment linear disposition model, with zero-order absorption for product use, on top of a mono-exponentially decreasing background. The background nicotine sub-model was introduced to account for the carry-over observed in single product use studies. As mentioned previously, the mono-exponential model assumed that product use occurred in the terminal phase of the preceding washout period. In retrospect, the t 1/2, 1 of 1.35 h supports this assumption. The typical terminal half-life t 1/2, z determined in the learning dataset was 17 h, supporting the previously observed carry-over after a washout period as long as 48 h. The previously reported value of approximately 2 h [8, 9] may better reflect the initial half-life t 1/2, 1 determined in the present work.
Some work already suggested a longer t 1/2, z of approximately 11 h [33]. This was estimated in urine samples, where the assay method sensitivity is higher than in plasma. While most reported nicotine analytical methods have a LLOQ from 0.5 to 1.0 ng/mL, the sensitivity of the analytical method in our studies was typically 0.2 ng/mL. Previous research on nicotine pharmacokinetics might have failed to capture the t 1/2, z probably due to a combination of a lower assay sensitivity and a shorter sampling period than used in our studies (24 h).
These results might have implications for future research, especially with regard to planned washout periods for clinical studies and the design of nicotine pharmacokinetic analyses. A t 1/2, z of 17 h warrants the prolongation of washout periods to prevent carry-over effects and to avoid the pitfalls of subtracting an observed baseline in case of non-compartmental pharmacokinetic analysis. Otherwise, for existing nicotine-containing products, one might select to deliberately shorten washout periods down to a duration that is deemed to be practicable, provided the data interpretation is model-based, and separates out the background exposure.
Influencing covariates were body weight, CYP2A6 activity, sex, presence of menthol, nicotine ISO yield, race (being black or not), and type of product.
Specifically, the bioavailability of nicotine for CC, NNS and nicotine gum relative to THS was 102, 24, and 61%, respectively, meaning that the corresponding background-adjusted exposure to nicotine was on average 2% higher, 76% lower and 39% lower than with THS, all other things being equal (i.e., subject characteristics and nicotine ISO yield). The absorption of nicotine from gum lasted 45 versus 5.3 min for the other products.
Owing to the unknown nicotine dose actually released by inhaled products, inter-individual variability (IIV) was assumed for Frel, producing the most significant drop in the −2LL statistic. The relative bioavailability decreased with increasing nicotine ISO yield and body weight. As smokers tend to self-titrate to their desired nicotine level, this might suggest that users tried to extract the same amount of nicotine from each product, resulting in high extraction efficiency from low nicotine-containing products, while a lower extraction was required from high nicotine-containing products. No clear-cut rationale was identified for the effect of weight on Frel.
Both sex and CYP2A6 activity influenced Cl/F. The sex-effect on Cl/F was apparently unrelated to sex-differences in weight, as weight itself was not found to impact Cl/F; therefore CYP2A6 activity at baseline was used as a marker of CYP2A6 polymorphism. A decrease in CYP2A6 activity in Japanese, Black and African Americans was expected, and a positive association between CYP2A6 activity and nicotine clearance is consistent with previously published data [8, 34, 35]. The effect of being black on C0 could not be distinguished from a study-effect, as all black or African American subjects in the learning dataset came from one study.
Menthol was related to small increases in V1/F and Tdur (9.5 and 5%, respectively) and there is no clear-cut explanation for this observation. It should be recognized, however, that the presence of menthol may be confounded by other factors, including region, product and product-use behavior. In fact, the nicotine gum was a menthol variant, which was only investigated in Japanese subjects in the present dataset. Menthol had no effect on Frel and Cl/F, which are the determinants of plasma nicotine exposure (AUC) at a given nicotine ISO yield.
Across the investigated dataset, background-adjusted exposure to nicotine was consistently lower with THS than with cigarettes, on average by 24 or 26%, depending on whether based on C max or AUC, respectively.
The evaluation of the final model was extended to an external validation dataset composed of 4 ad libitum use studies, representing 456 subjects and 4906 measurements, and comparable to the learning dataset with regard to demographic covariates. The external validation was conducted with all parameters fixed to the final estimates from the learning dataset except for C0, which was estimated from the validation dataset.
An inherent limitation of a population pharmacokinetic analysis is the dependency of the derived model on the analysis dataset. This raises the question as to whether our findings can be extrapolated to a different dataset of similar composition, to different nicotine-containing products, or to different populations. To some extent, the success of the external validation suggests that the model can accurately predict data outside the original dataset. This was feasible because design-related differences between the learning and validation datasets were clearly identified, and their translation in model terms did not require any change to the model structure. However, we speculate that extrapolation to different nicotine and tobacco products (e.g., transdermal systems) would require some adjustments to the model structure. Finally, extrapolating to populations not represented in the learning dataset would at least require changes to the covariate model.
In conclusion, the population pharmacokinetic model was able to describe the nicotine pharmacokinetics of a wide range of nicotine-containing products such as CC or THS, as well as of different nicotine replacement therapies (gum and NNS) with different routes of administration in different populations.
References
U.S Department of Health and Human Services. The health consequences of smoking: a report of the Surgeon General. Atlanta, GA: US Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health. 2004.
U.S Department of Health and Human Services. The health consequences of smoking—50 years of progress: a report of the Surgeon General. 2014.
Bilano V, Gilmour S, Moffiet T, d’Espaignet ET, Stevens GA, Commar A, et al. Global trends and projections for tobacco use, 1990–2025: an analysis of smoking indicators from the WHO Comprehensive Information Systems for Tobacco Control. Lancet. 2015;385(9972):966–76.
Unverdorben M, Mostert A, Munjal S, van der Bijl A, Potgieter L, Venter C, et al. Acute effects of cigarette smoking on pulmonary function. Regul Toxicol Pharmacol. 2010;57:241–6.
Roethig HJ, Koval T, Muhammad-Kah R, Jin Y, Mendes P, Unverdorben M. Short term effects of reduced exposure to cigarette smoke on white blood cells, platelets and red blood cells in adult cigarette smokers. Regul Toxicol Pharmacol. 2010;57(2–3):333–7.
Lüdicke F, Baker G, Magnette J, Picavet P, Weitkunat R. Reduced exposure to harmful and potentially harmful smoke constituents with the Tobacco Heating System 2.1. Nicotine Tob Res. 2017;19(2):168–75.
Picavet P, Haziza C, Lama N, Weitkunat R, Ludicke F. Comparison of the pharmacokinetics of nicotine following single and ad libitum use of a tobacco heating system or combustible cigarettes. Nicotine Tob Res. 2016;18(5):557–63.
Hukkanen J, Jacob P 3rd, Benowitz NL. Metabolism and disposition kinetics of nicotine. Pharmacol Rev. 2005;57(1):79–115.
Benowitz NL, Hukkanen J, Jacob P 3rd. Nicotine chemistry, metabolism, kinetics and biomarkers. Handb Exp Pharmacol. 2009;192:29–60.
Philip Morris Products S.A. A single-center, randomized, controlled, crossover study to investigate the nicotine pharmacokinetic profile and safety of THS 2.2 Menthol following single use in smokers compared to menthol conventional cigarettes and nicotine gum [ZRHM-PK-05-JP]. In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). NLM Identifier: NCT01967706. 2013–2014. http://clinicaltrials.gov/show/NCT01967706. Accessed 03 Jan 2016.
Philip Morris Products S.A. A single-center, open-label, randomized, controlled, crossover study to investigate the nicotine pharmacokinetic profile and safety of THS 2.2 following single use in smoking, healthy subjects compared to conventional cigarettes and nicotine gum [ZRHR-PK-02-JP]. In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). NLM Identifier: NCT01959607. 2013–2014. http://clinicaltrials.gov/show/NCT01959607. Accessed 26 Aug 2015.
Philip Morris Products S.A. A single-center, randomized, controlled, crossover study to investigate the nicotine pharmacokinetic profile and safety of THS 2.2 Menthol following single use in smokers compared to menthol conventional cigarettes and nicotine nasal spray [ZRHM-PK-06-US]. In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). NLM Identifier: NCT01967719. 2013–2014. http://clinicaltrials.gov/show/NCT01967719. Accessed 16 Jun 2015.
Philip Morris Products S.A. Nicotine pharmacokinetic profile and safety of the Tobacco Heating System 2.2 (THS 2.2) [ZRHR-PK-01-EU]. In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). NLM Identifier: NCT01967732. 2013–2014. http://clinicaltrials.gov/show/NCT01967732. Accessed 16 Jun 2015.
FDA (Food and Drug Administration). Guidance for industry: pharmacokinetics in patients with impaired renal function—Study design, data analysis, and impact on dosing and labeling-Draft guidance. 2010. http://www.fda.gov/downloads/Drugs//Guidances/UCM204959.pdf. Accessed 31 Jan 2017.
FDA (Food and Drug Administration). Guidance for industry: population pharmacokinetics. 1999. http://www.fda.gov/downloads/drugs/guidances/UCM072137.pdf. Accessed 31 Jan 2017.
Committee for Medicinal Products for Human Use (CHMP), European Medicines Agency (EMA). Guideline on reporting the results of population pharmacokinetic analyses (CHMP/EWP/185990/06). 2007. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003067.pdf. Accessed 31 Jan 2017.
Hooker AC, Staatz CE, Karlsson MO. Conditional weighted residuals (CWRES): a model diagnostic for the FOCE method. Pharm Res. 2007;24(12):2187–97.
Karlsson MO, Savic RM. Diagnosing model diagnostics. Clin Pharmacol Ther. 2007;82(1):17–20.
Hutmacher MM, Kowalski KG. Covariate selection in pharmacometric analyses: a review of methods. Br J Clin Pharmacol. 2015;79(1):132–47.
Ribbing J. Covariate model building in nonlinear mixed effects models. 2007; Uppsala; Acta Universatis Upsaliensis. Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Pharmacy 59. https://www.diva-portal.org/smash/get/diva2:170387/FULLTEXT01.pdf. Accessed 31 Jan 2017.
Wahlby U, Jonsson EN, Karlsson MO. Comparison of stepwise covariate model building strategies in population pharmacokinetic-pharmacodynamic analysis. AAPS PharmSci. 2002;4(4):E27.
Holford N. The visual predictive check–Superiority to standard diagnostic (Rorschach) plots. Poster presented at the annual meeting of the Population Approach Group in Europe, Pamplona, Spain. 2005. http://www.page-meeting.org/?abstract=738. Accessed 31 Jan 2017.
Levi M, Dempsey DA, Benowitz NL, Sheiner LB. Population pharmacokinetics of nicotine and its metabolites I. Model development. J Pharmacokinet Pharmacodyn. 2007;34(1):5–21.
Gupta SK, Benowitz NL, Jacob P 3rd, Rolf CN, Gorsline J. Bioavailability and absorption kinetics of nicotine following application of a transdermal system. Br J Clin Pharmacol. 1993;36(3):221–7.
Gabrielsson J, Weiner D. Pharmacokinetic and pharmacodynamic data analysis: concepts and applications. 4th ed. Stockholm: Swedish Pharmaceutical Press; 2006.
Le Houezec J, McNeill A, Britton J. Tobacco, nicotine and harm reduction. Drug Alcohol Rev. 2011;30(2):119–23.
Yildiz D. Nicotine, its metabolism and an overview of its biological effects. Toxicon. 2004;43(6):619–32.
Bergen AW, Michel M, Nishita D, Krasnow R, Javitz HS, Conneely KN, et al. Drug metabolizing enzyme and transporter gene variation, nicotine metabolism, prospective abstinence, and cigarette consumption. PLoS One. 2015;10(7):e0126113.
Robinson DE, Balter NJ, Schwartz SL. A physiologically based pharmacokinetic model for nicotine and cotinine in man. J Pharmacokinet Biopharm. 1992;20(6):591–609.
Teeguarden JG, Housand CJ, Smith JN, Hinderliter PM, Gunawan R, Timchalk CA. A multi-route model of nicotine-cotinine pharmacokinetics, pharmacodynamics and brain nicotinic acetylcholine receptor binding in humans. Regul Toxicol Pharmacol. 2013;65(1):12–28.
Plowchalk DR, Andersen ME, deBethizy JD. A physiologically based pharmacokinetic model for nicotine disposition in the Sprague-Dawley rat. Toxicol Appl Pharmacol. 1992;116(2):177–88.
Yamazaki H, Horiuchi K, Takano R, Nagano T, Shimizu M, Kitajima M, et al. Human blood concentrations of cotinine, a biomonitoring marker for tobacco smoke, extrapolated from nicotine metabolism in rats and humans and physiologically based pharmacokinetic modeling. Int J Environ Res Publ Health. 2010;7(9):3406–21.
Jacob P 3rd, Yu L, Shulgin AT, Benowitz NL. Minor tobacco alkaloids as biomarkers for tobacco use: comparison of users of cigarettes, smokeless tobacco, cigars, and pipes. Am J Publ Health. 1999;89(5):731–6.
Benowitz NL, Swan GE, Jacob P 3rd, Lessov-Schlaggar CN, Tyndale RF. CYP2A6 genotype and the metabolism and disposition kinetics of nicotine. Clin Pharmacol Ther. 2006;80(5):457–67.
Benowitz NL, Perez-Stable EJ, Herrera B, Jacob P 3rd. Slower metabolism and reduced intake of nicotine from cigarette smoking in Chinese–Americans. J Natl Cancer Inst. 2002;94(2):108–15.
Acknowledgements
The authors acknowledge the support of Valerie Poux, Philip Morris Products S.A., for editorial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Certara received funds from Philip Morris International for designing and conducting the analysis, and for writing the manuscript.
Conflict of interest
Author P. B. is a former employee of Philip Morris International. Authors N. L., R. W., and F. L. are employees of Philip Morris Product S.A.. Authors M. M. and H. M. are employees of Certara.
Ethical approval
The above mentioned model was built using the datasets of 8 clinical studies. All procedures performed in these studies involving human participants were conducted in accordance with the ethical standards of the institutional and national research committees and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Written informed consent was obtained from each subject who participated in one of the eight trials prior to performing any study-specific procedures.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Marchand, M., Brossard, P., Merdjan, H. et al. Nicotine Population Pharmacokinetics in Healthy Adult Smokers: A Retrospective Analysis. Eur J Drug Metab Pharmacokinet 42, 943–954 (2017). https://doi.org/10.1007/s13318-017-0405-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-017-0405-2