
Abstract
Background
Brain tissue hypoxia (PbtO2 < 20 mmHg) is common after subarachnoid hemorrhage (SAH) and associated with poor outcome. Recent data suggest that brain oxygen optimization is feasible and reduces the time spent with PbtO2 < 20 mmHg from 45 to 16% in patients with severe traumatic brain injury. Here, we intended to quantify the brain tissue hypoxia burden despite implementation of a protocolized treatment approach in poor-grade SAH patients and to identify the simultaneous occurrence of pathologic values potentially amenable to treatment.
Methods
We present a bi-centric observational cohort study including 100 poor-grade SAH patients admitted to two tertiary care centers who underwent multimodal brain monitoring and were managed with a PbtO2-targeted protocolized approach. PbtO2 optimization (≥ 20 mmHg) included a stepwise neuro-intensive care approach, aiming to prevent low cerebral perfusion pressure (CPP), and blood hemoglobin, and to keep normocapnia, normoxemia, and normothermia. Based on routine blood gas analysis, hemoglobin, PaCO2, and PaO2 data were matched to 2-h averaged data of continuous CPP, PbtO2, core temperature, and to hourly cerebral microdialysis (CMD) samples over the first 11 days.
Results
Patients had a Glasgow Coma Scale of 3 (IQR 3–4) and were 58 years old (IQR 48–66). Overall incidence of brain tissue hypoxia was 25%, which was not different between both sites despite differences in the treatment approach. During brain tissue hypoxia, episodes of CPP < 70 mmHg (27%), PaCO2 < 35 mmHg (19%), PaO2 < 80 mmHg (14%), Hb < 9 g/dL (11%), metabolic crisis (CMD-lactate/pyruvate ratio > 40, and CMD-glucose < 0.7 mmol/L; 7%), and temperature > 38.3 °C (4%) were common.
Conclusions
Our results demonstrate that brain tissue hypoxia remains common despite implementation of a PbtO2-targeted therapy in poor-grade SAH patients, suggesting room for further optimization.
Similar content being viewed by others
Introduction
Besides initial disease severity, secondary brain injury mechanisms largely contribute to the high mortality and morbidity after subarachnoid hemorrhage (SAH) [1]. Multimodal neuromonitoring may help to early identify tissue at risk which may be salvable using appropriate treatment strategies. In severe traumatic brain injury (TBI), a protocolized approach to increase brain tissue oxygen tension (PbtO2) has recently been shown to be feasible and significantly reduced the time of brain tissue hypoxia to 16% compared to the control group where PbtO2 < 20 mmHg occurred in 45% [2]. Although underpowered to detect an effect on outcome, the authors proved feasibility and descriptive outcome analysis seemed promising. Brain tissue hypoxia was managed by applying a hierarchical treatment algorithm including optimization of cerebral perfusion pressure (CPP), titration of pharmacologic analgesia, and sedation, adjustment of body temperature, optimization of oxygenation, targeting normocapnia, and red blood cell transfusions (RBC-transfusions) in anemic patients. Similar to severe TBI patients, prolonged brain tissue hypoxia was associated with poor outcome in poor-grade SAH patients [3]. Although PbtO2-based protocols were implemented in several intensive care units (ICUs), no prospective randomized study supports the use of such protocols. Moreover, it is not clear how high the hypoxic burden remains despite implementation of a protocolized treatment approach after SAH. Therefore, we aimed to analyze continuous neuromonitoring and systemic hemodynamic variables in two university centers using different treatment protocols to decrease episodes of brain tissue hypoxia.
In the current study, we intended to (1) assess the brain tissue hypoxia burden when a strict PbtO2-guided protocol is applied and (2) to identify factors that are concomitant to brain tissue hypoxia and may be amenable to modification in order to improve brain tissue hypoxia.
Methods
Study Design, Setting, and Participants
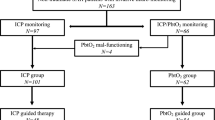
The study design was guided by the STROBE statement on observational cohort studies. Data of 105 consecutive patients admitted to the neurological ICU at two tertiary care centers (Medical University of Innsbruck = Neuro ICU [NICU] 1 and Medical University of Lausanne = NICU 2) diagnosed with non-traumatic SAH requiring multimodal neuromonitoring between 2010 and 2017 were prospectively collected. Five patients were excluded because of malfunctioning PbtO2 probes leaving 100 patients for final analysis. Inclusion criteria were (1) spontaneous SAH, (2) age ≥ 18 years, (3) multimodal neuromonitoring of intracranial pressure (ICP), and PbtO2, as part of routine clinical care. Invasive multimodal neuromonitoring was initiated in SAH patients requiring prolonged mechanical ventilation and/or clinical or radiological signs suggestive of increased intracranial pressure. The conduct of the study was approved by the ethics committee of the University of Innsbruck and Lausanne (Medical University Innsbruck, AN3898 285/4.8, AM4091-292/4.6; University of Lausanne, CER_VD 2016-01923). Written informed consent was obtained according to local regulations.
General Clinical Management and Grading
Initial disease severity was clinically quantified using the Glasgow Coma Scale (GCS) and World Federation of Neurological Surgeons (WFNS) Score. Standard treatment conformed to current international guidelines [4, 5]. Ruptured aneurysms were secured by clipping or endovascular coiling. All patients were mechanically ventilated, received appropriate sedation and were continuously monitored for mean arterial pressure (MAP). Prophylactic parenteral (NICU 1) and oral (NICU 2) nimodipine were administered in all patients. Transcranial color-coded duplex sonography (TCD) was routinely obtained in order to follow patients for vasospasm. Vasospasm was defined as elevation of mean velocities > 120 cm/s in the middle or anterior cerebral artery or daily change in mean TCD-velocities > 50 cm/s. Severe vasospasm (> 200 cm/s) was further confirmed by cerebral angiography and treated after interdisciplinary discussion using intra-arterial nimodipine. Delayed cerebral ischemia (DCI) was defined as the occurrence of a new focal neurologic deficit, a decrease of at least 2 points on the Glasgow Coma Scale or a new infarct on the computed tomography (CT) or magnetic resonance imaging scan not attributable to other causes [6]. Functional outcome was evaluated at 3 months post-bleeding by a study nurse blinded to monitor data with the modified Rankin Scale Score (mRS) in NICU 1. In NICU 2, functional outcome was evaluated 6 months after the onset of SAH using the Glasgow Outcome Score (GOS). Outcome was categorized into good (mRS 0–2, GOS 4–5) and poor outcome (mRS 3–6, GOS 1–3).
Data Collection and Multimodal Neuromonitoring
Baseline characteristics, demographics, hospital complications, and outcomes were prospectively recorded in the institutional SAH databases. Physiologic variables were continuously recorded using the patient data management system (NICU 1: PDMS, CentricityTM Critical Care 8.1 SP2; GE Healthcare Information Technology, Dornstadt German; NICU 2: MetaVision, iMDsoft, Düsseldorf, Germany). Based on clinical and radiological criteria, patients underwent intracranial neuromonitoring including measurement of PbtO2, ICP, and cerebral metabolites. Neuromonitoring probes were inserted into the hemisphere deemed to be at greatest risk of secondary brain injury either through a frontal burr hole using a triple-lumen bolt or tunneled and placed in the white matter. Probe location was confirmed by CT-scans obtained within 24 h of implantations. In NICU 1, probe location was defined as perilesional in the presence of a focal hypodense or hyperdense lesion within a 1-cm radius of the tip of the PbtO2 probe, intralesional or otherwise as normal-appearing healthy brain tissue. In NICU 2, all PbtO2 probes were in healthy appearing brain tissue assessed on head CT-scans. PbtO2 was measured using Licox® CC1.SB probes (NICU 1: Integra LifeSciences, Ratingen, Germany; NICU 2: Integra Neurosciences, Plainsboro, NJ), ICP by an intraparenchymal probe (NICU 1: Neurovent-P-temp, Raumedic®, Helmbrechts, Germany; NICU 2: ICP Codman®, Raynham, MA). CPP was calculated by MAP, measured at the level of the foramen of Monro, minus ICP. Cerebral microdialysis (CMD) was performed using a 20 or 100 kD-cutoff-microdialysis catheter (CMA 70 or CMA-71; µDialysis, Stockholm, Sweden). The CMD catheter was perfused with artificial sterile cerebrospinal fluid (Isotonic; Perfusion Fluid CNS; µDialysis AB, Stockholm, Sweden) at a rate of 0.3 µL/min. Hourly samples were immediately analyzed at the bedside for CMD-lactate, CMD-pyruvate, CMD-glucose, and CMD-glutamate using a point-of-care analyzer (ISCUSflex; µDialysis AB, Sweden) and frozen at − 80 °C.
Management of Brain Oxygen
Brain tissue hypoxia (PbtO2 < 20 mmHg for greater than 10 min) was treated according to institutional protocols (Fig. 1). Treatment options were left to the discretion of the treating neuro-intensivists including optimization of CPP ≥ 70 mmHg with vasopressors if necessary and fluid administration to maintain euvolemia, RBC-transfusions in anemic patients (NICU 1 goal Hb ≥ 8 g/dL, NICU 2 goal Hb ≥ 9 g/dL integrating PbtO2) and targeting normocapnia (PaCO2 ≥ 35 mmHg), normothermia (36.5–37.5 °C) using targeted temperature management, optimization of oxygenation (PaO2 ≥ 80 mmHg), and titration of analgesia, and sedation.

Institutional protocols to treat brain tissue hypoxia (PbtO2 < 20 mmHg) of NICU 1 and NICU 2
Outcome
The primary outcome parameter of the current study was the incidence of brain tissue hypoxia within the study period. Brain tissue hypoxia was defined as PbtO2 < 20 mmHg [7].
Data Management and Statistical Analysis
In order to account for the association of hemoglobin and blood gases and PbtO2, continuous data of CPP, PbtO2, and core temperature as well as hourly measured CMD-samples were averaged over 2 preceding hours and matched to hemoglobin, PaO2, and PaCO2 levels derived from routine blood gas analysis. The study period was defined as the first 11 days with the day of admission until midnight denoted as day 0. PbtO2 values were checked for plausibility and manually cleaned for artifacts, which resulted in exclusion of 5.6% of monitored PbtO2 data. Hb-levels were categorized into four ranges that approximated the quartiles of their distribution: < 9, 9–10, 10–11, > 11 g/dL. Based on the published literature [2], we predefined variables which potentially qualify for interventions to prevent episodes of brain tissue hypoxia and calculated the incidence during episodes of brain tissue hypoxia over the first 11 monitoring days: CPP < 70 mmHg, anemia (Hb < 9 g/dL), metabolic crisis (CMD-lactate/pyruvate ratio [LPR] > 40 along with neuroglucopenia [CMD-glucose < 0.7 mmol/L]) [8], fever (core temperature > 38.3 °C), PaCO2 < 35 mmHg, and PaO2 < 80 mmHg.
Continuous variables were assessed for normality and reported as mean ± standard error of mean or median and interquartile range (IQR). Categorical variables were reported as counts and proportions in each group. Groups were compared in univariate analysis using the t test, Mann–Whitney U test or Fisher’s exact test, as appropriate. Brain tissue hypoxia was evaluated as dichotomized as well as continuous variable.
Univariate and/or multivariable generalized estimating equation models were used for all analysis of repeated measurements [9]; with co-variates specified in the results section. Cases with missing values were included. Five patients who were lost to follow-up were excluded from functional outcome analysis.
All analyses and graphical representations were performed with IBM-SPSS V24.0 (SPSS Inc., Chicago, IL, USA) and Prism 5 for Windows V 5.01 (GraphPad Software, Inc., LA Jolla, CA 92037 USA). A p value < 0.05 was set as statistically significant threshold.
Results
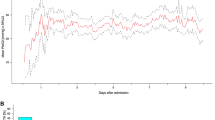
A total of 100 consecutive poor-grade SAH patients were studied. Patients’ baseline characteristics and hospital complications at each site are detailed in Table 1. Altogether, 927 neuromonitoring days with median 11 (IQR 9–11) days per patient were analyzed. This resulted in 5841 analyzed PbtO2 matched blood gas samples with a median of 7 samples (IQR 4–10) per patient days. No clinically significant complication attributable to PbtO2 probe insertion occurred. In NICU 1, multimodal monitoring placement associated bleeding was observed in 3/66 (4.5%) patients. However, none of these bleedings was directly associated with the brain tissue oxygen probe. In 5/66 (7.5%) patients, the PbtO2 probe was contaminated with gram-positive rods without any signs of meningitis, encephalitis or brain abscess formation. Overall mean PbtO2 was 26 ± 0.1 mmHg and increased over time from 25 ± 0.6 mmHg (day 1) to 28 ± 0.5 mmHg on day 8 (p = 0.1, Fig. 2). In 75% of the study time, the targeted goal of PbtO2 ≥ 20 mmHg was successfully achieved. Episodes of brain tissue hypoxia despite protocolized PbtO2 treatment occurred in 81% of patients and 25% of analyzed time episodes. The incidence of brain tissue hypoxia was the highest on day 1 (31%) and the lowest on day 9 (20%) (p = 0.047). Assessment of predefined concomitant abnormal values revealed low CPP (< 70 mmHg) as most common abnormal factor during episodes of brain tissue hypoxia (27%) followed by PaCO2 < 35 mmHg (19%), PaO2 < 80 mmHg (14%), Hb < 9 g/dL (11%), metabolic crisis (7%), and temperature > 38.3 °C (4%) (Fig. 3). During episodes of brain tissue hypoxia, CPP < 70 mmHg was most commonly found on day 1 compared to the rest of the monitoring time (p = 0.01, Fig. 2). Inversely, incidence of anemia significantly increased over time (p < 0.001, Fig. 2). Other potentially treatable factors did not significantly change over time.

Mean (± SEM) PbtO2-levels over time are shown. Percentage of anemia (Hb < 9 g/dL) increased, whereas low cerebral perfusion pressure (CPP)-levels (< 70 mmHg) decreased during episodes of brain tissue hypoxia (PbtO2 < 20 mmHg) over the study period

Bars represent the percentage of simultaneous abnormal values shown in the x-axis during the time of brain tissue hypoxia (PbtO2 < 20 mmHg). CPP cerebral perfusion pressure, Hb hemoglobin
Absolute mean PbtO2 levels decreased from 25 ± 0.6 mmHg on day 1 to 23 ± 0.6 mmHg on day 5 in patients with DCI and increased to a maximum of 28 ± 0.8 mmHg on day 8 secondary to induced hypertension with CPP ≥ 70 mmHg (days 6–10: mean 82 ± 0.4 mmHg). Day-wise comparisons of absolute PbtO2 values showed significantly lower PbtO2 values in patients with vasospasm on days 2–6 (p < 0.001). Similarly, PbtO2 values were significantly lower in patients with DCI as compared to those without DCI on days 3–6 (p < 0.01, Supplementary Fig. 1). Reactive to therapeutic interventions, PbtO2 increased to a higher level as compared to baseline. Interestingly, we did not find a lower incidence of predefined abnormal values during episodes of normal PbtO2 (≥ 20 mmHg) as compared to episodes of brain tissue hypoxia.
In NICU 1, 57% of PbtO2 probes were placed in healthy tissue with overall mean PbtO2 values in normal range (26 ± 0.19 mmHg), perilesional probe location was recorded in 26% of patients with similar mean PbtO2 values (27 ± 0.45 mmHg, p = 0.473) and intralesional probe location was evident in 7% of patients with significantly lower mean PbtO2 values (18 ± 0.59 mmHg, p < 0.001). In the remaining patients, head CT-scan revealed global cerebral edema (mean 29 ± 0.56 mmHg).
No association was found between PbtO2-levels and poor functional outcome after 3 and 6 months (adjOR 0.98/mmHg, 95% CI 0.94–1.02, p = 0.32) independently of established outcome parameters (WFNS grade, loss of consciousness, age, and anemia, Hb < 9 g/dL) in 95 patients.
Site-Specific Differences
Patients’ demographics were comparable across the two sites; however, patients admitted to NICU 2 had a lower GCS, a higher mFisher score, and a higher DCI rate (Table 1). Institutional protocols to target PbtO2-levels above 20 mmHg differed between NICU 1 and NICU 2 (Fig. 1). In NICU 1, interventions to treat brain tissue hypoxia were used without a hierarchical order, whereas a stepwise approach was followed in NICU 2. Moreover, NICU 2 targeted Hb-levels ≥ 9 g/dL and in NICU 1, the goal was ≥ 8 g/dL. Twenty-nine percent of patients were transfused during the first 11 days (NICU 1: 27%, NICU 2: 32%; p = 0.65). Mean pre-transfusion hemoglobin levels among transfused patients were higher in NICU 2 (8.6 ± 0.1 g/dL) as compared to NICU 1 (7.8 ± 0.2 g/dL; p = 0.001), whereas mean nadir hemoglobin levels in non-transfused patients were 9.1 ± 0.2 g/dL (NICU 1: 8.5 ± 0.2, NICU 2: 10.6 ± 0.3; p < 0.001). Similarly, patients in NICU 2 had significantly higher CPP-levels as compared to patients treated in NICU 1 (p < 0.001, Fig. 4). In this line, mean-2 h-CPP < 70 mmHg during episodes of brain tissue hypoxia (< 20 mmHg) was lower in NICU 2 (NICU 1: 35%, NICU 2: 8%; p < 0.001). Overall brain hypoxic episodes (PbtO2 < 20 mmHg for greater than 10 min) requiring treatment interventions occurred in 32% (NICU 1: 33%, NICU 2: 29%; p = 0.5). Within each patient, the median percentage of PbtO2 < 20 mmHg (> 10 min) during the study period was 21% (IQR 9–56%) (NICU 1: 24%, IQR 10–73; NICU 2: 19%, IQR 2–44; p = 0.09). Day-wise comparison revealed a significantly lower incidence of brain tissue hypoxia in NICU 2 as compared to NICU 1 only on day 1 (p = 0.01). Importantly, during DCI-risk time, there was no significant center specific difference in achieving normal PbtO2-levels despite significantly higher CPP-levels in NICU 2 (Fig. 4). Comparable site-specific differences were obtained for the mean daily AUC (area under curve) of brain tissue hypoxia and time spent in brain tissue hypoxia (Fig. 5).

Center-specific mean (± SEM) cerebral perfusion pressure (CPP)-levels and frequencies of brain tissue hypoxia (PbtO2 < 20 mmHg for at least 10 min) over the study period. *p < 0.05; **p < 0.01; ***p < 0.001

Brain tissue hypoxia (PbtO2 < 20 mmHg) burden at each site based on PbtO2 mean values of 5-min intervals. a The daily brain tissue hypoxia burden, defined as the mean (± SEM) area under the curve of brain tissue hypoxia (= sum of depth of abnormalities multiplied by the time spent in PbtO2 < 20 mmHg normalized to monitored time) is reported in mmHg*minutes. NICU 1 is represented by the darker shades of gray, NICU 2 by the lighter shades of gray. b Time spent in brain tissue hypoxia expressed in daily mean (± SEM) minutes (normalized to monitored time). NICU 1 is represented by the darker shades of gray, NICU 2 by the lighter shades of gray. c Average depth of brain tissue hypoxia (the mean (± SEM) of the PbtO2 values < 20 mmHg normalized to monitoring time). NICU 1 is represented by the darker shades of gray, NICU 2 by the lighter shades of gray
Discussion
In this study, we provide clinical data of protocolized PbtO2-guided therapy to prevent brain tissue hypoxia in poor-grade SAH patients in two independent university centers. In the majority of analyzed time periods (75%), the goal of PbtO2 ≥ 20 mmHg was successfully achieved. Still, our data suggest that further optimization of systemic variables may be needed. Protocols used in our institutions aimed at optimization of CPP, hemoglobin levels, and sedation depth and maintaining normocapnia, normothermia, and euvolemia which are potential interventions to improve PbtO2 [2].
Among these factors potentially resulting in low PbtO2 levels, CPP-values below 70 mmHg were detected most often. In this line, previous studies found an association between CPP < 70 mmHg and a significant higher incidence of brain tissue hypoxia in poor-grade SAH patients [10]. Interventions to increase CPP include hemodynamic augmentation by maintaining euvolemia or induced hypertension using vasopressors [4, 5]. Importantly, we found a lower incidence of episodes with CPP < 70 mmHg during the time when DCI occurred underlining the current recommendation of induced hypertension in patients with DCI. Interestingly, CPP-levels were higher in NICU 2 but did not result in a lower incidence of brain tissue hypoxia during DCI-risk time. In contrast, higher CPP-levels in the initial phase after SAH were related to a higher brain tissue oxygenation in NICU 2. This is of interest and reflects the complex interaction of these variables which are influenced by cerebral autoregulatory capacity (oxygen reactivity index, Orx) [11], neurovascular coupling, CO2 reactivity [12], and other factors resulting in increased consumption of local PbtO2, including spreading depolarizations [13], fever, and seizures. Based on our data, we cannot support the idea of further CPP augmentation without a multimodal neuromonitoring approach in the delayed phase after SAH. Even more interesting, episodes of CPP < 70 mmHg were also found in 25% of time with normal PbtO2-levels. This may simply reflect that clinicians at both institutions used a PbtO2-based protocol and accepted lower CPP-values when PbtO2 levels were within normal range. Finally, it may indicate that de-escalation of CPP augmentation was performed as long as brain tissue hypoxia did not occur.
Delivery of oxygen to the brain tissue is further diminished by hyperventilation which is associated with vasoconstriction of cerebral blood vessels. This is important, since all patients were sedated and mechanically ventilated during multimodal neuromonitoring time. Therefore, PaCO2-levels can easily be adjusted by ventilator settings targeting normocapnia or hypercapnia in case of normal ICP. This is consistent with previous findings, suggesting that hyperventilation is frequent after severe brain injury [14] and associated with increased risk for brain tissue hypoxia [15]. In contrast, controlled hypercapnia improved cerebral blood flow and brain oxygenation in a prospective trial [16].
We found anemia (< 9 g/dL) during episodes of brain tissue hypoxia in 11% of observation time. Anemia is a common complication following SAH and has been independently associated with poor outcome [17]. Moreover, an association between anemia and brain tissue hypoxia has been described [18, 19]. RBC-transfusions resulted in increased delivery of oxygen in around 75% of interventions [20, 21]. Some trials suggest improved outcome in patients targeting higher hemoglobin levels [17, 22]. However, the use of RBC-transfusion to correct anemia is controversial due to its potential risks and association with increased morbidity [23]. It is important to mention that site-specific differences with higher Hb-thresholds in NICU 2 were not associated with higher PbtO2-levels.
We also identified energy metabolic crisis during episodes of brain tissue hypoxia. Elevated CMD-lactate-to-pyruvate-ratio (LPR) together with low CMD-glucose levels in the presence of ischemia suggest increased anaerobic metabolism [24]. This may occur during failure of sufficient energy delivery or increased demand. On the other hand, a high LPR may also result from mitochondrial dysfunction of non-ischemic etiology [25]. Importantly, brain metabolic distress was linked to higher hospital mortality in severely brain-injured patients [26].
Similarly, an increase in body and brain temperature increases brain energy demand. Fever is a common complication after SAH [27, 28] and linked to poor functional outcome [29, 30]. Fever has further been associated with secondary brain injury including vasospasm [31] and DCI [32]. Interestingly, fever was linked to higher LPR-levels indicative of higher cerebral metabolism and presumably increased oxygen consumption in TBI patients [33]. Targeted temperature management with aggressive fever control to achieve normothermia was implemented in both centers resulting in a low overall incidence of fever (6%).
Abnormal modifiable factors during episodes of brain tissue hypoxia may be a target for further improvement in clinical practice. However, treatment adaptations have to be thoroughly considered following an individualized concept in each patient. The natural history of disease (e.g., progressive anemia, DCI) as well as the complex reasons for brain tissue hypoxia is of special importance when using a protocolized approach. This bi-centric study with comparable effects among the two sites despite different treatment protocols suggests generalizability to other institutions following a similar protocol.
Limitations
Optimization of brain tissue oxygenation is targeted to avoid secondary brain injury including DCI. Furthermore, low PbtO2-levels may be associated with poor outcome [3]. In our analysis, brain tissue hypoxia was not independently predictive of poor functional outcome. One explanation may be that both centers followed a PbtO2-guided therapy and a control group were therefore lacking. Second, the dataset was based on blood gas analysis and matched to mean 2-h PbtO2-levels with limited numbers of analyzed PbtO2-samples. However, evaluation of PbtO2 during the whole neuromonitoring time in NICU 1 did not reveal a higher incidence of brain tissue hypoxia and lower PbtO2-levels were not independently associated with poor outcome (data not shown). The study is further limited by varying daily measurements of Hb-levels within patients (IQR 4–10). An overrepresentation of patients with multiple blood gas analyses per day is possible. However, disease severity did not differ in these patients and PbtO2-levels were similar (data not shown). The definition of anemia as hemoglobin levels < 9 g/dL irrespective of sex and protocol used is another limitation. However, severely brain-injured patients might have some benefit of higher transfusion thresholds, which is currently investigated in the TRAIN study (ClinicalTrials.gov Identifier: NCT02968654). Moreover, the approach of comparing two centers implicates a certain degree of heterogeneity of how measurements are obtained. Finally, we present a retrospective analysis of prospectively recorded data. Therefore, we cannot provide detailed information about the effectiveness of interventions used to increase PbtO2.
Conclusions
This bi-center observational cohort analysis demonstrates that despite the implementation of a PbtO2-guided therapy, brain tissue hypoxia still occurred during 25% of neuromonitoring time. We found that low CPP, hypocapnia, low PaO2, anemia, metabolic crisis, and fever were frequent during brain tissue hypoxia making them to potential targets for interventions to prevent brain tissue hypoxia. Despite applying a protocolized PbtO2 treatment approach, we could not replicate the previously described association between brain tissue hypoxia and poor outcome after SAH.
Abbreviations
- SAH:
-
Subarachnoid hemorrhage
- TBI:
-
Traumatic brain injury
- PbtO2 :
-
Brain tissue oxygen tension
- CPP:
-
Cerebral perfusion pressure
- RBC-transfusion:
-
Red blood cell transfusion
- ICP:
-
Intracranial pressure
- MAP:
-
Mean arterial pressure
- CMD:
-
Cerebral microdialysis
References
Nieuwkamp DJ, Setz LE, Algra A, et al. Changes in case fatality of aneurysmal subarachnoid haemorrhage over time, according to age, sex, and region: a meta-analysis. Lancet Neurol. 2009;8(7):635–42.
Okonkwo DO, Shutter LA, Moore C, et al. Brain oxygen optimization in severe traumatic brain injury phase-II: a phase II randomized trial. Crit Care Med. 2017;45(11):1907–14.
Kett-White R, Hutchinson PJ, Al-Rawi PG, et al. Adverse cerebral events detected after subarachnoid hemorrhage using brain oxygen and microdialysis probes. Neurosurgery. 2002;50(6):1213–21; discussion 21-2.
Connolly ES, Rabinstein AA, Carhuapoma JR, et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/american Stroke Association. Stroke. 2012;43(6):1711–37.
Steiner T, Juvela S, Unterberg A, et al. European Stroke Organization guidelines for the management of intracranial aneurysms and subarachnoid haemorrhage. Cerebrovasc Dis. 2013;35(2):93–112.
Vergouwen MD, Vermeulen M, van Gijn J, et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke. 2010;41(10):2391–5.
Stiefel MF, Spiotta A, Gracias VH, et al. Reduced mortality rate in patients with severe traumatic brain injury treated with brain tissue oxygen monitoring. J Neurosurg. 2005;103(5):805–11.
Helbok R, Kofler M, Schiefecker AJ, et al. Clinical use of cerebral microdialysis in patients with aneurysmal subarachnoid hemorrhage-state of the art. Front Neurol. 2017;8:565.
Chan JSK. Analysis of correlation structures using generalized estimating equation approach for longitudinal binary data. J Data Sci. 2014;12:293–305.
Schmidt JM, Ko SB, Helbok R, et al. Cerebral perfusion pressure thresholds for brain tissue hypoxia and metabolic crisis after poor-grade subarachnoid hemorrhage. Stroke. 2011;42(5):1351–6.
Jaeger M, Schuhmann MU, Soehle M, Nagel C, Meixensberger J. Continuous monitoring of cerebrovascular autoregulation after subarachnoid hemorrhage by brain tissue oxygen pressure reactivity and its relation to delayed cerebral infarction. Stroke. 2007;38(3):981–6.
Balbi M, Koide M, Wellman GC, Plesnila N. Inversion of neurovascular coupling after subarachnoid hemorrhage in vivo. J Cereb Blood Flow Metab. 2017;37(11):3625–34.
Dreier JP, Woitzik J, Fabricius M, et al. Delayed ischemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations. Brain. 2006;129(Pt 12):3224–37.
Williamson CA, Sheehan KM, Tipirneni R, et al. The association between spontaneous hyperventilation, delayed cerebral ischemia, and poor neurological outcome in patients with subarachnoid hemorrhage. Neurocrit Care. 2015;23(3):330–8.
Coles JP, Fryer TD, Coleman MR, et al. Hyperventilation following head injury: effect on ischemic burden and cerebral oxidative metabolism. Crit Care Med. 2007;35(2):568–78.
Westermaier T, Stetter C, Kunze E, et al. Controlled hypercapnia enhances cerebral blood flow and brain tissue oxygenation after aneurysmal subarachnoid hemorrhage: results of a phase 1 study. Neurocrit Care. 2016;25(2):205–14.
Naidech AM, Drescher J, Ault ML, et al. Higher hemoglobin is associated with less cerebral infarction, poor outcome, and death after subarachnoid hemorrhage. Neurosurgery. 2006;59(4):775–9; discussion 9-80.
Kurtz P, Schmidt JM, Claassen J, et al. Anemia is associated with metabolic distress and brain tissue hypoxia after subarachnoid hemorrhage. Neurocrit Care. 2010;13(1):10–6.
Oddo M, Milby A, Chen I, et al. Hemoglobin concentration and cerebral metabolism in patients with aneurysmal subarachnoid hemorrhage. Stroke. 2009;40(4):1275–81.
Kurtz P, Helbok R, Claassen J, et al. The effect of packed red blood cell transfusion on cerebral oxygenation and metabolism after subarachnoid hemorrhage. Neurocrit Care. 2016;24(1):118–21.
Dhar R, Zazulia AR, Derdeyn CP, Diringer MN. RBC transfusion improves cerebral oxygen delivery in subarachnoid hemorrhage. Crit Care Med. 2017;45(4):653–9.
Naidech AM, Jovanovic B, Wartenberg KE, et al. Higher hemoglobin is associated with improved outcome after subarachnoid hemorrhage. Crit Care Med. 2007;35(10):2383–9.
Kramer AH, Roberts DJ, Zygun DA. Optimal glycemic control in neurocritical care patients: a systematic review and meta-analysis. Crit Care. 2012;16(5):R203.
Hlatky R, Valadka AB, Goodman JC, Contant CF, Robertson CS. Patterns of energy substrates during ischemia measured in the brain by microdialysis. J Neurotrauma. 2004;21(7):894–906.
Jacobsen A, Nielsen TH, Nilsson O, Schalen W, Nordstrom CH. Bedside diagnosis of mitochondrial dysfunction in aneurysmal subarachnoid hemorrhage. Acta Neurol Scand. 2014;130(3):156–63.
Oddo M, Schmidt JM, Carrera E, et al. Impact of tight glycemic control on cerebral glucose metabolism after severe brain injury: a microdialysis study. Crit Care Med. 2008;36(12):3233–8.
Wartenberg KE, Schmidt JM, Claassen J, et al. Impact of medical complications on outcome after subarachnoid hemorrhage. Crit Care Med. 2006;34(3):617–23; quiz 24.
Kilpatrick MM, Lowry DW, Firlik AD, Yonas H, Marion DW. Hyperthermia in the neurosurgical intensive care unit. Neurosurgery. 2000;47(4):850–5; discussion 5-6.
Rincon F, Patel U, Schorr C, et al. Brain injury as a risk factor for fever upon admission to the intensive care unit and association with in-hospital case fatality: a matched cohort study. J Intensive Care Med. 2015;30(2):107–14.
Fernandez A, Schmidt JM, Claassen J, et al. Fever after subarachnoid hemorrhage: risk factors and impact on outcome. Neurology. 2007;68(13):1013–9.
Oliveira-Filho J, Ezzeddine MA, Segal AZ, et al. Fever in subarachnoid hemorrhage: relationship to vasospasm and outcome. Neurology. 2001;56(10):1299–304.
Douds GL, Tadzong B, Agarwal AD, et al. Influence of Fever and hospital-acquired infection on the incidence of delayed neurological deficit and poor outcome after aneurysmal subarachnoid hemorrhage. Neurol Res Int. 2012;2012:479865.
Oddo M, Frangos S, Milby A, et al. Induced normothermia attenuates cerebral metabolic distress in patients with aneurysmal subarachnoid hemorrhage and refractory Fever. Stroke. 2009;40(5):1913–6.
Acknowledgements
Open access funding provided by Austrian Science Fund (FWF). We would like to thank Adriano Bernini and Anna Lindner for data acquisition. Moreover, we would like to thank the nursing staff and the entire team of both intensive care units for their ongoing contribution in the care of our patients.
Funding
This study has received funding from the Austrian Science Fund (FWF) under project number KLI 375 (to RB).
Author information
Authors and Affiliations
Contributions
VR and RH were involved in the study idea and design, data acquisition and analysis, writing and drafting the manuscript. DS, MG, BI, MK, AS, JPM, PM, RB, BP, and MO were involved in the study idea, data acquisition and drafting the manuscript. CT was involved in the study idea and design as well as drafting the manuscript. All authors read and approved this version of the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors report no conflict of interest concerning the materials or methods used in this study or the findings specified in this paper.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed Consent
Informed consent was obtained from all individual participants included in the study according to local regulations.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
12028_2019_753_MOESM1_ESM.tif
Supplementary Figure 1: Mean (± SEM) PbtO2 values and frequencies of brain tissue hypoxia (PbtO2 < 20 mmHg) in patients with and without DCI (delayed cerebral ischemia) over the study period. **p < 0.01, ***p < 0.001 (TIFF 644 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Rass, V., Solari, D., Ianosi, B. et al. Protocolized Brain Oxygen Optimization in Subarachnoid Hemorrhage. Neurocrit Care 31, 263–272 (2019). https://doi.org/10.1007/s12028-019-00753-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12028-019-00753-0