
Abstract
A novel fluorescent dye bis-(pyridin-2-yl-methyl)-(1,3,4-triphenyl-1H-pyrazolo[3,4-b]quinolin-6-ylmethyl)-amine (P1) has been synthesized and investigated by means of steady state and time-resolved fluorescence techniques. This compound acts as sensor for fluorescence detection of small inorganic cations (lithium, sodium, barium, magnesium, calcium, and zinc) in highly polar solvents such as acetonitrile. The mechanism which allows application of this compound as sensor is an electron transfer from the electron-donative part of molecule (amine) to the acceptor part (pyrazoloquinoline derivative), which is retarded upon complexation of the electro-donative part by inorganic cations. The binding constants are strongly dependent on the charge density of the analyzed cations. The 2/1 complexes of P1 with Zn++ and Mg++ cations posses large binding constants. Moreover, in the presence of these cations a significant bathochromic shift of fluorescence is observed. The most probable explanation of such behaviour is the formation of intramolecular excimer. This is partially supported by the quantum chemical calculations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Molecular systems based on 1,3,4-triphenyl1H-pyrazolo[3,4-b]quinoline fluorophore were found to be good sensors [1] for small inorganic cations also in less polar solvents as tetrahydrofuran. Such compounds are attractive for fluorescence detection of nonfluorescent analytes such as metal ions (sodium, lithium, calcium, barium, etc.) in biological systems [2, 3], These cations are involved in many biological processes of fundamental importance for life and monitoring of these ions occurring in small amounts in blood and urine is of major importance in medicine. Typical concentrations of these cations in blood and urine (data in parantheses) are 143 (125) mM, 5 (65) mM, 1 (4) mM, and 1.5 (4) mM for Na+, K+, Mg++ and Ca++, respectively [2]. Concentration of the lithium cation in plasma is estimated as 0.09–0.15 μmol/l [4]. On the other hand, barium salts are known as toxic agents (classic symptoms of barium toxicity are repeated profound hypokalemia, cardiac arrhythmias, respiratory failure, prolonged gastrointestinal dysfunction, paralysis, myoclonus, hypertension, and profound lactic acidosis) [5], the maximal concentrations of this cation in serum is in range 3–20 μg/dl [6].
In the last years much attention has been paid to developing fluorescence sensors.
The working principle of the previously investigated systems is as follows. The molecules which contain electron donor and acceptor moieties (so called EDA systems) are linked by the methine subunit. The donor part of the molecule is the called recognition moiety. In the ground state these two parts are almost non-interactive. Upon excitation to the first singlet state an effective transfer of one electron from the donative part of the molecule to the azaromatic skeleton occurs. In consequence the fluorescence of the dye is strongly quenched. However, the recognition moiety (electron donor part of the molecule) can act in the ground state as a Lewis base. Thus, upon addition of the Lewis acid to the EDA system a complex is formed and the electro-donative property of this part of molecule is strongly diminished. Therefore, quenching of the fluorescence is weaker in the presence of cations and the system emits the fluorescence light. In consequence, the fluorescence intensity depends on the cation concentration, making these compounds usable for qualitative determination of the analyzed cations.
It should be noted that the working principle for some other molecular systems might be different than that presented above. There is for instance a group of sensors containing polycyclic aromatic hydrocarbon fluorophores (like naphthalene or pyrene) which are linked by crown ether or open-chain structure (podand). In such compounds addition of the cations, that are then bounded to the azacrown moiety, changes the ratio of the excimer-monomer fluorescence quantum yields [7–9].
It has been established that in many cases the indicators undergo 1/1 and 2/1 (ligand/metal ion) complexation [10–12]. Thus, the following equilibria should be taken into account to describe the overall complexation process.
and
In our paper we would like to discuss the fluorometric data on a novel compound being derivative of 1,3,4-triphenyl-1H-pyrazolo[3,4-b]quinoline.
Experimental
The Synthesis of P1 Molecule
The synthesis of chelate is depicted in the Scheme 1. 6-methyl-1,3,4-triphenyl-1H-pyrazolo[3,4-b]quinoline 1 was prepared by reaction of equimolar amounts p-toluidine, benzaldehyde and 2,5-diphenyl-2,4-dihydropyrazol-3-one according to literature procedures [13]. The bromination of this compound with NBS yielded 6-bromomethyl-1,3,4-triphenyl-1H-pyrazolo[3,4-b]quinoline 2 [1]. The nucleophilic substitution of bromomethyl group with di-(2-picolyl)amine in acetonitrile gave compound 3.

a NBS/CCl4 ; b di-(2-picolyl)amine/K2CO3/AcCN.
Bis-(pyridin-2-yl-methyl)-(1,3,4-triphenyl-1H-pyrazolo[3,4-b]quinolin-6-ylmethyl)-amine 3
6-Bromomethyl-1,3,4-triphenyl-1H-pyrazolo[3,4-b]quinoline 2 (490 mg, 1 mmol), potassium carbonate (140 mg, 1 mmol) and di-(2-picolyl)amine were refluxed together in acetonitrile (10 ml) for 2 hours. The solution was filtered and evaporated. The oily residue was dissolved in chloroform and filtered through a short pad of alumina. The product was chromatographed on column packed with alumina and eluted with toluene:acetone mixture (9:1).
Yellow powder, 300 mg, 50%, mp 205–206 °C.
1H NMR(300 MHz, CDCl3, δppm): 8.55–8.53(m, 2H); 8.44–8.42(m, 2H); 8.15(d, J = 8.8 Hz, 1H, 8-H); 7.88(s, 1H, 5-H); 7.81(dd, J = 8.8 Hz, 1.9 Hz, 1H, 7-H); 7.57–7.50(m, 4H); 7.40–7.00(m, 16 H); 3.75(s, 4H); 3.74(s, 2H).
13C NMR(75 MHz, CDCl3, δppm):160.28; 150.87; 149.58; 148.83; 147.36; 144.73; 140.57; 137.00; 135.85; 135.33; 133.26; 132.36; 131.05; 129.78; 129.68; 128.95; 128.53; 128.19; 128.13; 126.61; 126.02; 123.89; 123.32; 122.57; 121.55; 115.60; 60.69; 59.11.
Anal. calcd for C41H32N6: C 80.90; H 5.30; N 13.80. C 80.78; H 5.16; N 13.56.
MS (ES): (m/z): 609.2 (M+) , 410.2, 332.1.
Melting points were determined on a Mel-Temp Apparatus II (capillary). NMR spectra were recorded on a Varian (Mercury) spectrometer at 25 °C.
The ab initio calculations were performed using DFT(B3LYP) method for the ground state and time dependent perturbation method TD-DFT(B3LYP) for the excited state with the cc-pVDZ basis set. Computations were performed with the Gaussian09 program. The optimised geometry of the ground state was calculated for the investigated ligand and its complexes with Zn++ and Mg++ cation.
Fluorescence
The solvents: cyclohexane (CHX), dibutyl ether (DBE), butyl chloride (BuCl), ethyl acetate (EtAc), tetrahydrofuran (THF), dichloromethane (CH2Cl2), methanol (MeOH), acetonitrile (ACN), dimethylformamide (DMF), and dimethyl sulfoxide (DMSO) were of spectroscopic grade and were used as received (all from Aldrich). All the solvents did not show any traces of fluorescence. For fluorescence the solutions of the dye were degassed using multiple freeze-pump-thaw cycles. The sample concentration of the dyes for spectroscopic measurements was ca. 10−5 M (this corresponds to absorbance of ca. 0.2–0.3 at the excitation wavelengths used for the fluorescence investigations). Lithium, sodium, barium, magnesium, calcium (tetrahydrate) and zinc (hexahydrate) perchlorate (Aldrich) were used as received. In an independent measurement it was found that a small addition of deionised water couldn’t influence significantly the fluorescence of the acetonitrilic solution of the dye. Fluorescence titration experiments using added salts are performed without degassing procedure.
Fluorescence measurements were made on a home-built spectrofluorimeter and a time-correlated single photon counting setup. For time-resolved fluorescence measurements a picosecond diode laser (λ = 400 nm, 70 ps pulse duration) (IBH-UK) was used as the excitation source. For steady-state fluorescence measurements a 365 or 405-nm line of medium-pressure mercury lamp was used. The fluorescence quantum yield measurements were carried out with quinine sulphate in water (Φ fl = 0.55) [14] as an actinometer.
The equilibrium constants (K 1) for complexation of the ligand by alkali cations are calculated by fitting the Eq. 1 to the relative fluorescence intensities in the presence of different concentration (cM) of the employed alkaline metal perchlorates [15].
where Φ(0), Φ(∞) and Φ(cM) is the fluorescence intensity of the dye alone, with the large concentration of the salt and with the cM concentration of the salt, respectively, c L indicates the analytical concentration of the ligand.
For 2/1 complexation the binding constant K 2 is defined by the Eq. 2 [1]:
The Eq. 1 can be converted into more complex one:
where x denotes the concentration of the complex \( ML_2^{+} \). This quantity can be obtained by numerical solving of the third degree polynomial:
with the following parameters:
The values of the binding constants K 1 and K 2 were obtained from fluorescence intensities using nonlinear least-square fitting coded in FORTRAN.
Results
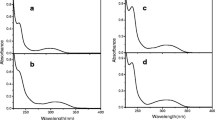
Absorption spectra of the studied dye are rather insensitive to solvent polarity. Addition of the studied perchlorate salts affects the absorption only in the UV spectral range as pointed out in Fig. 1.

Absorption spectra of P1 in acetonitrile without (solid) and with 0.01 M LiClO4 (dotted) and 0.01 M Zn (ClO4)2 (dashed)

Dependence of fluorescence lifetimes of P1 (top), fluorescence quantum yields (top inset) and nonradiative rate constant (bottom) on the solvent dielectric constants. The correlation lines are drawn only for presentation purposes.
Fluorescence quantum yields of the dyes depend strongly on solvent polarity. In solvents of low polarities the fluorescence quantum yields are very high and decrease significantly with increasing solvent polarity, even though the position of the maximum is almost not influenced by polarity of the solvent.
The fluorescence decay functions are clearly monoexponential in all employed solvents. In solvents possessing dielectric constants less that 21 (i.e. less polar than acetone) the fluorescence lifetimes are rather long and close to the value characteristic of the parent molecule 1,3,4-triphenyl1H-pyrazolo[3,4-b]quinoline (ca 17 ns). This indicates that the intramolecular electron transfer, responsible for the fluorescence quenching, is not operative in solvents of low and medium polarities. The situation changes in highly polar solvents such as acetonitrile. In these solvents an effective quenching is observed. This causes a shortening of the fluorescence lifetimes and quantum yields as can be recognized from the data collected in Table 1.
The dependence of these parameters on the solvent dielectric constants are presented in Fig. 2.
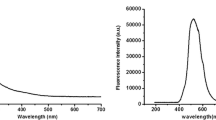
Upon addition of perchlorates to the acetonitrile solutions of the dye P1 the fluorescence significantly increases. The position of the fluorescence maximum is not changed due to added salts except in the presence of Zn++ and Mg++. The dependence of the fluorescence quantum yield on the salt concentration is presented in Fig. 3.

Salt concentration dependence (in logarithmic scale) of the relative fluorescence quantum yield of P1 upon addition of lithium (black circles), sodium (open diamonds), calcium (black squares), barium (black triangles), magnesium (open circles) and zinc perchlorates (stars) in acetonitrile. The correlation lines are calculated either from eq. 1 (Li+, Na+, Ba++ and Ca++) or eqs 2–3 (Mg++ and Zn++).
The binding constants calculated using eqs. 1 and 2-3 are collected in Table 2.
Discussion
The studied molecule does not show a significant solvent polarity influence as regards both the ground state absorption band and the position of the fluorescence maximum. However, the recognition moiety (1-(pyridin-2-yl)-N-(pyridin-2-ylmethyl)methanamine) influences significantly the fate of the fluorescing state of the fluorophore. Fluorescence lifetimes and quantum yields depend on solvent polarity as can be predicted from the electron transfer theory. In the excited state one electron from the donative part of the molecule is transferred to the acceptor part, forming the nonfluorescing monomolecular radical pair which recombines recovering the ground state of the system. Increasing of the solvent polarity decreases the energy of the charge transfer pair, which is formed in monomolecular charge transfer process, resulting in more efficient fluorescence quenching in polar solvents. In solvents of low and medium polarity the quenching is hardly observed but already in acetone the significant reduction of these parameters is observed. These observations may be useful in designing this molecule as a fluorescence indicator of small inorganic cations.
The change of the first absorption band upon addition of the salts is not significant. Only small influence in the deep UV-region is observed (cf. Fig. 1). Addition of small amounts (up to 1×10−1 M) of the perchlorates (lithium, sodium, calcium, barium, magnesium and zincum) to the solution of P1 in acetonitrile causes a ca. 4–7-fold increase of the fluorescence intensity. It means that even a small concentration of the cations is sufficient to complexate the dye by the cation and this complex is stable in the excited singlet state. It implies a large sensitivity and good selectivity of this dye to the presence of the investigated cations as can be recognized from the different binding constants (Tab. 2). Thus, this dye may be used as a fluorescing indicator for the cations in acetonitrile.
An interesting behaviour has been observed upon addition of the bivalent cations like Mg++ and Zn++. Contrary to the systems investigated previously (K1 and L1) [1] addition of these cations causes a significant change of the spectral distribution in the fluorescence of the formed complexes as presented in Fig. 4.

Fluorescence spectra of P1 (solid line) and that of 2/1 complexes of P1 with Zn (dotted line) and Mg (dashed line) in acetonitrile.
These complexes have a stoichometry 2/1 as can be judged from the salt concentration dependence of the integral intensity of the fluorescence. The question which remains to be addressed is to explain the reason of the spectral shift of the fluorescence band in the presence of Zn and Mg cations. We postulate that the factor responsible for the small fluorescence shift in the presence of these cations is a formation of an intramolecular excimer- like product.
The red shift in the fluorescence spectrum of P1 in the presence of Zn++ in acetonitrile is about 17 nm, while the half-with of the fluorescence band is enlarged by ca. 15 nm (94 vs 109 nm). Similar changes (although less pronounced) occur in the presence of Mg++. The fluorescence lifetime of the complex P1 with Zn++ measured at different observation wavelengths (i.e. at the blue and red side of the fluorescence spectrum) differ one from another but the difference is rather small (10.7 vs 12.1 ns).
Excimer Formation
If the excited molecule (M*) undergoes a collision with the same but not excited molecule (M) an excited dimer is formed (MM)*. The formation of the bimolecular excimers is a diffusional process, therefore the formation is strongly preferable at higher molecule concentrations because this process proceeds within the fluorescence lifetime of the molecule.
It is well-known that many aromatic and planar molecules (such as naphthalene and anthracene) form excimers. This manifests itself by existence of the additional, broad and structureless fluorescence band of MM* located at wavelengths higher than that of monomer M*. In some cases these bands are well resolved. The intensity of the excimer fluorescence grows up when the concentration of M increases. In such excimers the time evolution of the fluorescence of the monomer and the excimer are different. The decay of the monomer fluorescence (M*) is the sum of two exponents whereas in the case of the excimer fluorescence (MM*) the preexponential factors have opposite signs. It means that the rise and the decay of the excimer fluorescence is observed.
When two monomers are linked by a flexible chain the intramolecular excimer may be also formed [18, 19], Formation of the excimer involves not a translational motion but a rotational one to form the sandwich-type structure. When the ground state complex (MM) already exists then displacement of the two molecules to achieve a sandwich configuration appropriate for the excimer fluorescence occurs very quickly. In such cases the rise time of the excimer fluorescence could not be detected. Such type excimers, where an excimer emission appears or disappears in the presence of ions, are commonly used as fluorescing ion indicators [20].
To check the possibility of existence of the excimer-like form in our systems we decided to perform some theoretical investigations on our systems and the excimer consisting of two simpler 1H-pyrazolo[3,4-b]quinoline molecules (PQ). This molecule resembles P1 but contains smaller number of atoms which renders the quantum chemical calculations in the excited state possible.
Quantum Chemical Calculations
The computed vertical excitation energy for the ligand molecule (P1) is 2.87 eV and the relaxed fluorescence energy is 2.44 eV (Table 3). These values are very close to excitation energies for the bare fluorophore (P2) without the recognition unit (1-(pyridin-2-yl)-N-(pyridin-2-ylmethyl)methanamine), proving that this group does not take part in the excitation. Large size of the investigated complexes causes that the vertical excitation energies and the energy of fluorescence from the relaxed excited state could be calculated only for the single fluorophore (P1). The fluorescence spectral shift caused by the excimer formation between ligands of 2:1 complexes were estimated by performing computations for a simpler model molecule. For this purpose, 1H-pyrazolo[3,4-b]quinoline molecule (PQ) was chosen as the fluorophore and its excimer consisting of two PQ molecules was examined (see Scheme 2).

Chemical structures of the molecules characterized in Table 3. Top—PQ (1H-pyrazolo[3,4-b]quinoline), bottom P2 (6-methyl 1,3,4-triphenyl-1H-pyrazolo[3,4-b]quinoline).
The influence of the phenyl groups on the spectroscopic properties of the system was investigated by comparison with 6-methyl 1,3,4-triphenyl-1H-pyrazolo[3,4-b]quinoline (P2). Although the presence of amino group (in P1) causes the existence of the CT state, the charge transfer cannot occur if the nitrogen atom lone pair is used for complexation with a metal cation. Thus, the neglected substituents should not modify the spectroscopic properties of the complex.
The ground state geometry of the dimer PQ·PQ was optimised for the distance between monomers of ca. 10.5 Å to 11.8 Å (the NN’ distances) where the distance between the relevant nitrogen atoms of both molecules is larger by 0.7 Å than the distance between the carbon atoms at the opposite end of the molecules. Thus, this system may be treated as consisting of isolated molecules. The optimised geometry of the excited state, however, points out to the excimer formation with much smaller equilibrium distance (R eq) between monomers equal ca. 3.4 Å (CC′ = 3.385 Å, NN′ = 3.498 Å, see Fig. 5).

PQ·PQ* excimer. Lines mark distances: CC′ = 3.385 Å and NN′ = 3.498 Å
The relaxed fluorescence energy for the excimer PQ·PQ* is 2.47 eV while for the isolated monomer the energy 3.17 eV is predicted. This strong red-shift is as large as −0.7 eV and it overwhelms the experimental fluorescence red-shift which is only −0.09 eV for the P12Zn++ complex and −0.06 eV for the complex with Mg++ cation. The above results show that the fluorescence red-shift could be extraordinarily large if the PQ molecules would interact optimally and both molecular planes were close to each other. This is certainly not the case of our systems P12Zn++ and P12Mg++.
The optimised structures of the complexes in the ground state are shown in Fig. 6. The most important effect of the substituent groups (phenyl rings) which should be here taken into account is the sterical hindrance that they cause. The phenyl substituents at positions 3 and 4 in the ground state are twisted with respect to the fluorophore plane by 46 and 68 degrees in, respectively. These angles are expected to decrease in the excited state but only slightly. Thus, the optimal distance between the phenyl rings of the two monomers of about 3.4 Å causes that the fluorophores could not approach closer each other than 4.8 Å.

Optimised structures of the P12Zn++ (bottom) and P12Mg++ (top) in the ground state
The excimer formation in the complexes P12Zn++ and P12Mg++ is facilitated by conformational change concerning primarily the rotation of the fluorophore groups around the Namino-CH2 bonds to make the long axis of the fluorophores parallel and the rotation with respect to the CH2-fluorophore bonds to align the fluorophores in nearly parallel planes. The possible conformation of the P12Zn++ excimer is presented in Fig. 7. Thus, the minimal distance between the 1,3,4-triphenyl1H-pyrazolo[3,4-b]quinoline fluorophores in the real excimer is limited by the minimal distance between the two carbon atoms of the -CH2- bridges linking the recognition moiety (1-(pyridin-2-yl)-N-(pyridin-2-ylmethyl)methanamine) with the 1,3,4-triphenyl1H-pyrazolo[3,4-b]quinoline. The Mg-Namino and Zn-Namino bond length in the ground state are equal 2.30 Å. Taking into account the linking N-CH2 bonds, the parts of the fluorophores connected to this carbon atoms should not approach each other closer than roughly 5.5 Å if the core complex and the -CH2- bridges are assumed to be rigid. This rather large separation between the fluorophore’s planes seems to be responsible for only small bathochromic shift of the excimer fluorescence as compared to that for the bare P1 molecule.

Hypothetical (unoptimised) structure of excimer P12Zn++* obtained by rotation around the N-CH2 bonds linking the fluorophores with the core complex
To illustrate the dependence of the spectral shift of fluorescence on the distance between the fluorophores the excitation energy curve was calculated (see Fig. 8) for PQ·PQ*. The open circles mark the fluorescence energy calculated for the excimer geometries obtained from the optimised geometry by increasing the distance between the monomers without reoptimisation. Therefore this curve is relevant for distances close to the minimum energy point but at large distances the calculation predicts too high energies and false limit for isolated fluorophores. To overcome this deficiency and to give a more reliable estimation of the fluorescence red-shift at the distances available for the P12Zn++ and P12Mg++ excimers the geometries of PQ·PQ* were reoptimised for frozen few values of the CC′ distance marked in Fig. 5. The CC′ distances and the fluorescence energies are collected in Tab.3 and graphically presented in Fig. 8 (full circles). A good agreement with the experimental red-shift for the P12Zn++ complex (ca. 0.08 eV) is achieved at the distance roughly 5 Å. At this distance the value of the fluorescence energy for the excimer PQ.PQ* is equal 3.09 eV while for the isolated molecule PQ* it is 3.17 eV . For smaller distances the fluorescence energy decreases rapidly. On the other hand, for distances larger than 5.5 Å the fluorescence energy is essentially the same as for the isolated fluorophore. Finally, it should be noticed that the computations with larger basis sets and with correction for Van der Waals dispersion interaction could predict an effective excimer formation and a significant red-shift fluorescence at slightly larger distances than the TD-DFT(B3LYP)/cc-pVDZ predictions discussed above.

Distance dependence of the fluorescence energies of the PQ·PQ* excimer. Open circles mark the energy of the excimer calculated for frozen geometry of monomers taken from the equilibrium structure (distance 3.385 Å), full circles mark energy of the optimised excimer. For more detailed explanation see text above
Conclusions
A novel fluorescent sensor for determination of small inorganic cations was synthesized and investigated by means of fluorescence spectroscopy and quantum-chemical calculations. The stability of the fluorescent complexes of P1 with inorganic cations (Mn+) (P1Mn+ and P12M n+) were determined by fluorescence titration. This indicator shows a qualitatively good selectivity for small inorganic cations and good sensitivity especially towards bivalent cations such as Zn++ and Mg++. This system may be applicative in practice for determination of these cations in the presence of other ones.
The enhanced sensitivity and selectivity for Zn++ and Mg++ arises from an efficient complexation of these cations with two P1 molecules.
Moreover, the remarkable bathochromic shift in fluorescence (17 nm for P12Zn++ and 13 nm for P12Mg++ complex) is observed. This spectral feature is most probably due to excimer formation. This is partially supported by the quantum chemical calculations.
References
Mac M, Uchacz T, Wróbel T, Danel A, Kulig E (2010) J Fluoresc 20:525
Valeur B (2002) Molecular fluorescence. Principles and applications. Ch. 10. Wiley-VCH, Weinheim
Li YQ, Bricks JL, Resch-Genger U, Spieles M, Rettig W (2006) J Phys Chem A 110:10972
Leflon P, Plaquet R, Rose F, Hennon G, Ledeme N (1996) Anal Chim Acta 327:301
Jacobs IA, Taddeo J, Kelly K, Valenziano C (2002) Am J Ind Med 41:285
Hung Y-M, Chung H-M (2004) Nephrol Dial Transplant 19:1308
Bergamini G, Ceroni P, Balzani V, Cornelissen L, van Heyst J, Lee S-K, Vögtle F (2005) J Mater Chem 15:2959
Licchelli M, Orbelli Biroli A, Poggi A, Sacchi D, Sangermani C, Zema M (2003) Dalton Trans 4537
Bouas-Laurent H, Castellan A, Daney M, Desvergne JP, Guinand G, Marsau P, Riffaud MH (1986) J Am Chem Soc 108:315
Yamauchi A, Hayashita T, Nishizawa S, Watanabe M, Teramae N (1999) J Am Chem Soc 121:2319
Xia WS, Shmehl RH, Li CJ (1999) J Am Chem Soc 121:5599
Leray I, Habib-Jiwan J-L, Branger C, Soumillion J-P, Valeur B (2000) J Photochem Photobiol A, Chem 135:163
Tomasik P, Chaczatrian K, Chaczatrian G, Danel A (2003) Pol J Chem 77:1141
Meech SR, Phillips D (1983) J Photochem 23:193
Bourson J, Pouget J, Valeur B (1993) J Phys Chem 97:4552
Barkici H, Koner AL, Nau WM (2005) Chem Commun 5411.
Leray I, Lefevre J-P, Delouis J-F, Delaire J, Valuer B (2001) Chem Eur 7:4590
Lewis FD, Wu T, Burch EL, Bassani DM, Yang J-S, Schneider S, Jager W, Letsinger RL (1997) J Am Chem Soc 119:5451
Yuasa H, Miyagawa N, Izumi T, Nakatani M, Izumi M, Hashimoto H (2004) Org Lett 6:1489
Lee SH, Kim SH, Kim SK, Jung JH, Kim JS (2005) J Org Chem 70:9288
Acknowledgments
The authors would like to thank Dr Andrzej M. Turek for editorial comments. Quantum chemical calculations were carried out in the Academic Computer Center “Cyfronet” in Krakow, Poland (supported by Grant No. MNiSW/IBM_BC_HS21/UJ/075/2009).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Mac, M., Uchacz, T., Danel, A. et al. A New Fluorescent Sensor Based on 1H-pyrazolo[3,4-b]quinoline Skeleton. Part 2. J Fluoresc 21, 375–383 (2011). https://doi.org/10.1007/s10895-010-0726-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10895-010-0726-x