
Abstract
Plectin is a large, 500-kDa, intermediate filament (IF)-associated protein. It acts as a cytoskeletal crosslinker and signaling scaffold, affecting mechanical as well as dynamic properties of the cytoskeleton. As a member of the plakin family of cytolinker proteins, plectin has a multidomain structure that is responsible for its vast binding portfolio. It not only binds to all types of IFs, actin filaments and microtubules, but also to transmembrane receptors, proteins of the subplasma membrane protein skeleton, components of the nuclear envelope, and several kinases with known roles in migration, proliferation, and energy metabolism of cells. Due to alternative splicing, plectin is expressed as various isoforms with differing N-terminal heads that dictate their differential subcellular targeting. Through specific interactions with other proteins at their target sites and their ability to bind to all types of IFs, plectin molecules provide strategically located IF anchorage sites within the cytoplasm of cells. In this review, we will present an overview of the structural features and functional properties of plectin and discuss recent progress in defining the role of its isoforms in stress-prone tissues and the implicated diseases, with focus on skin, skeletal muscle, and Schwann cells of peripheral nerve.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The cytolinker protein plectin, although able to interact with actin filaments and microtubules, is primarily an intermediate filament (IF)-binding protein. In this capacity, it anchors IFs to strategic cellular sites such as focal adhesions, desmosomes and hemidesmosomes (HDs), Z-disks and costameres, intercalated disks, as well as the nuclear, mitochondrial, and subplasma membrane skeleton, conferring mechanical stability to the IF network and thereby to the cell. Structurally, plectin bears no similarity to IF proteins except for possessing a central, in this case unusually long, α-helical coiled coil rod domain that mediates dimerization and higher oligomer formation and is instrumental for networking and bridging different cellular structures. As plectin is expressed in practically all cell types, plectin mutations result in a pleiotropic phenotype affecting simultaneously several tissues primarily skin, muscle, and nerve.
Plectin, an IF-associated protein
Plectin was originally isolated from IF-enriched preparations of rat glioma C6 cells (Pytela and Wiche 1980). A few years later, a protein then named 300 K intermediate filament-associated protein (IFAP-300 K) was isolated from baby hamster kidney (BHK-21) cells based on its ability to codistribute with vimentin IFs (Lieska et al. 1985). In 1992, a protein referred to as HD-1 was identified as a major component of HDs isolated from bovine corneal epithelial cells (Hieda et al. 1992). With the advent of molecular cloning and sequencing, it was possible to show that the three proteins were identical (Clubb et al. 2000; Okumura et al. 1999; Wiche et al. 1991). In addition to its copurification and colocalization with IFs, plectin has been shown to directly bind to several IF subunit proteins including vimentin, glial fibrillary acid protein, neurofilaments (Foisner et al. 1988), desmin (Reipert et al. 1999), type I and II keratins (Geerts et al. 1999; Steinböck et al. 2000), and lamin B (Foisner et al. 1991). There is evidence that plectin-IF interactions are modulated by phosphorylation (Herrmann and Wiche 1983, 1987). For example, plectin dissociates from vimentin upon phosphorylation by protein kinase C (PKC), whereas binding to vimentin is enhanced by cAMP-dependent protein kinase (PKA) phosphorylation (Foisner et al. 1991). Likewise, plectin’s association with lamin B is significantly decreased upon PKA- and PKC-phosphorylation of either binding partner (Foisner et al. 1991). During mitosis, plectin is phosphorylated by the cyclin-dependent kinase CDK1, triggering its dissociation from vimentin (Foisner et al. 1996). As the various plectin isoforms allocate IF-docking sites to different subcellular locations, plectin molecules exert a significant influence over the spatial and temporal distribution of IF networks (Wiche and Winter 2011; and see below).
Plectin: more than a crosslinker of the cytoskeleton
Besides its association with IFs, plectin has been shown to interact with the other two major cytoskeletal filament systems, actin-based microfilaments, and microtubules (MTs). Plectin interaction with the microfilament system was demonstrated by ultrastructural studies using immunogold-labeled antibodies to plectin. When whole mount electron microscopy was applied to glioma C6 cells, plectin was detected as thin (3-nm) filamentous structures that linked IFs (vimentin) to each other and also to actin filaments (Foisner et al. 1995). This association is consistent with plectin’s occurrence at microfilament–plasma membrane junctions in all types of muscle (Wiche et al. 1983), its buildup at focal adhesions, and its colocalization with actin stress fibers (Andrä et al. 1998). Furthermore, plectin has a functional actin-binding domain (ABD; Andrä et al. 1998) that is structurally similar to those of the spectrin/dystrophin superfamily of actin-binding proteins (Fontao et al. 2001, García-Alvarez et al. 2003, Sevcík et al. 2004). The binding affinity of plectin for actin is comparable to that of other actin-binding proteins (Andrä et al. 1998) such as dystonin (Yang et al. 1996), tensin (Lo et al. 1994), and dystrophin (Jarrett and Foster 1995). The involvement and impact of plectin linking IFs to focal adhesions has recently been established (Bhattacharya et al. 2009; Burgstaller et al. 2010).
Plectin has been shown to bind to microtubule-associated proteins (MAPs; Herrmann and Wiche 1987), to copurify with MTs through repeated cycles of in vitro assembly/disassembly (Koszka et al. 1985), to form cross-bridges between IFs and MTs in fibroblasts depleted of the actin cytoskeleton (Svitkina et al. 1996), and to be part of the MT interactome during cell division (Gache et al. 2010; Mack and Compton 2001; Sauer et al. 2005). In a very recent report, it was shown that plectin binds to MAP2 via its SH3 domain. By seizing MAP2, plectin antagonizes MAP-mediated MT stabilization, and thus, destabilizes MTs (Valencia et al. 2013).
As plectin deficiency favors stress fiber formation and reduces MT dynamics, the binding of plectin to microfilaments and MTs cannot merely be seen as providing a structural scaffold but rather as contributing to their dynamic regulation.
Gene structure: alternative splicing as a source of functional diversity
Plectin has been cloned from a rat glioma C6 cell cDNA library (Wiche et al. 1991), a human placenta cDNA library (Liu et al. 1996), and a mouse skeletal muscle cDNA library (Fuchs et al. 1999). Likewise, plectin’s gene locus has been analyzed in human (Liu et al. 1996; McLean et al. 1996), rat (Elliott et al. 1997), and mouse (Fuchs et al. 1999) genomes. The human plectin gene was mapped to 8q24 at the end of the long arm of chromosome 8 (Liu et al. 1996; McLean et al. 1996), and the mouse gene to a syntenic region on chromosome 15 (Fuchs et al. 1999); the rat gene is located on chromosome 7 (RefGene, GeneID: 64204). The gene spans over 62 kb and is split into 41 exons. The most striking feature is that multiple variable first exons are spliced into a common exon 2 (Fig. 1a, b). Nine of these exons (1-1g, 1k) are first coding exons (Fuchs et al. 1999; McInroy and Määttä 2011), while the coding regions of three others (1h-1j) start in exon 6, generating variants lacking most of plectin’s (exons 2–8 encoded) ABD (Rezniczek et al. 2003). Downstream of exon 2 there are 31 mostly constant exons, with exons 2–8, encoding the ABD (Liu et al. 1996; Sevcik et al. 2004). Two additional small exons, 2α and 3α, within the ABD-encoding genomic region are optionally spliced within exons 2 and 4. The following 22 exons, exons 9–30, encode the plakin domain (see below) of plectin, whereas single large exons (31 and 32) encode the central and C-terminal domains, respectively. Low level expression of a variant-lacking rod-encoding exon 31 has also been reported (Elliott et al. 1997; Fuchs et al. 2005).

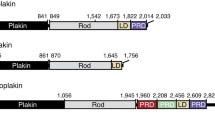
Schematic representation of the plectin gene and the encoded protein. a Genomic organization of the plectin gene. Variable first coding exons are represented by colored boxes, constant exons by black boxes, introns by lines, and splicing by diagonal lines with the color code as for the exons; arrows indicate direction of transcription. b Transcripts generated by alternative splicing of the 5′ end of the gene. Exons are indicated by boxes, orange areas within the boxes denote noncoding regions, black areas, coding regions. Brown boxes indicate coding sequences if preceded by one of the first coding exons (1-1 g, 1 k), or noncoding sequences if preceded by one of the noncoding exons (1 h-1j). The black area within exon 6 denotes the first coding region of transcripts starting with exons 1 h, 1i, and 1j. Two optionally spliced exons inserted between exons 2 and 4 are shown in red. c Accession numbers of plectin transcript variants and isoforms. d Schematic representation of the protein. The N-terminal domain contains an actin-binding domain (ABD) consisting of two calponin homology domains (dark and light red), and a plakin domain comprising nine spectrin repeats (green) and one SH3 domain (yellow). The central rod domain is a 200-nm-long coiled coil. The C-terminal domain contains six plectin repeat domains, each consisting of a conserved region, referred to as module (blue), and a linker region, one of which harbors the IF-binding domain (IFBD, light blue). The different N termini of the isoforms are indicated by a star. Positions of the mapped binding regions to various proteins are indicated below the scheme
The particular structure of the plectin gene has unveiled the mechanism for the generation of transcript variants differing only in their first coding exon, as each of the 12 variable exons is separately spliced into a common set of downstream exons. A similar gene structure has been found only in a few other genes (Zhang et al. 2004). Thus, most plectin isoforms differ just in their short N-terminal sequences (Fig. 1b). These short sequences are of crucial importance, however, as they determine the cellular localization of the isoforms and confer their tissue-specific expression (Rezniczek et al. 2003). Plectin isoform 1 (P1), for example, is targeted to the nucleus/ER membrane (Rezniczek et al. 2003), plectin 1a (P1a) to HDs (Andrä et al. 2003), P1b to mitochondria (Winter et al. 2008), P1c to MTs (Valencia et al. 2013), P1d to Z-disks (Konieczny et al. 2008), and P1f to focal adhesions and costameres (Burgstaller et al. 2010, Konieczny et al. 2008).
Expression and cellular localization
Plectin is expressed in a wide variety of tissues and cell types, and it is particularly abundant in tissues subjected to great mechanical stress, such as stratified and simple epithelia, skeletal and heart muscle, and blood vessels (Wiche et al. 1983, 1984). At the cellular level, plectin is found at plasma membrane attachment sites of IFs and microfilaments, for instance, Z-disks in stratified muscle, dense plaques in smooth muscle, intercalated disks in cardiac muscle, HDs in the basal cell layer of stratified epithelia, desmosomes, and focal adhesions. Furthermore, plectin accumulates in cells forming tissue layers at the interface between tissues and fluid-filled cavities such as kidney glomeruli, liver bile canaliculi, bladder urothelium, gut villi, ependymal cell layers lining the cavities of brain and spinal cord, gial-endothelial interfaces, and endothelial cells of blood vessels (Errante et al. 1994; Lie et al. 1998; Wiche et al. 1983; Yaoita et al. 1996). Plectin is also a major component of trabecular meshwork cells of the eye which control intraocular pressure (Inoue et al. 2010).
Protein structure and binding partners
Full-length plectin is expressed as a 499–533-kDa protein depending on the particular plectin isoform. Using rotary shadowing electron microscopy, plectin has been visualized as a dumbbell-like structure comprising a central 200-nm-long rod domain flanked by two large globular domains (Foisner and Wiche 1987). This structure has been confirmed by secondary structure predictions based on plectin’s amino acid sequence (Wiche et al. 1991). Functionally, plectin is a multidomain protein with similarities to other cytoskeletal proteins of the plakin family (Sonnenberg and Liem 2007). The amino-terminal domain harbors a conventional ABD preceding the plakin domain (Fig. 1d). The ABD is composed of two calponin homology domains closely resembling the ABD of fimbrin (Sevcík et al. 2004), whereas the plakin domain comprises nine spectrin repeats with one Src-homology 3 (SH3) domain inserted in repeat 5 (Sonnenberg et al. 2007). The central rod domain contains an almost continuous 1127 residue long, mainly α-helical coiled coil showing long stretches of heptads repeats with a staggered charge periodicity of 10.4 for both acidic and basic residues (Wiche et al. 1991). The C-terminal domain contains six plectin repeat domains (PRDs) and a short terminal tail. Each PRD is built up of a conserved core region and short linker sequences connecting the cores (Janda et al. 2001, Wiche et al. 1991). The PRDs are most likely tightly packed through interlinkage by hydrogen bonds and disulfide bridges (Spurny et al. 2007), forming a compact C-terminal globular domain. The C-terminal tail represents a 70 residues long serine-rich region.
As a protein composed of multiple functional domains, plectin is able to interact with a vast array of different proteins via one or more of those domains (Figs. 1d, 2). Thereby it can act as a mechanical reinforcement or a scaffold for signaling molecules, or both. Plectin binds to vimentin (Nikolic et al. 1996), desmin (Favre et al. 2011; Reipert et al. 1999), glial fibrillary acidic protein (Foisner et al. 1988), lamin B (Foisner et al. 1991), and cytokeratins (Steinböck et al. 2000) via an IF-binding site located in the linker region between PRDs 5 and 6. The IF-binding site has been further narrowed down to a stretch of ~50 amino acids containing a basic residue cluster typically found in nuclear localization signals (Nikolic et al. 1996). The ABD of plectin turned out to be multifunctional, binding not only to actin (Fuchs et al. 1999) but also to integrin β4 via the first pair of fibronectin type III repeats (Geerts et al. 1999; Rezniczek et al. 1998), vimentin (Sevcik et al. 2004), nesprin 3 (Wilhelmsen et al. 2005), the EF-ZZ domain of dystrophin and utrophin (Rezniczek et al. 2007), and calmodulin (Kostan et al. 2009). Binding of F-actin and integrin β4 to the ABD of plectin has been shown to be mutually exclusive (Geerts et al. 1999; Litjens et al. 2003). Additional binding sites for integrin β4 and β-dystroglycan have been mapped to the plakin and C-terminal repeat domains of plectin (Koster et al. 2004; Rezniczek et al. 1998, 2007). The plakin domain also serves as a binding site for the hemidesmosomal transmembrane protein, bullous pemphigoid antigen 180 (BPAG2; Koster et al. 2003), and the nonreceptor tyrosine kinase Fer (Lunter and Wiche 2002). Furthermore, the SH3 domain embedded in the plakin domain serves as a docking site for MAP2 (Valencia et al. 2013). Regarding the rod domain, there is evidence that it mediates plectin self-association via coiled coil interactions (Foisner and Wiche 1987; Walko et al. 2011). It also recruits caspase 8 that cleaves plectin toward the end of the rod (Asp 2395) in the early stages of apoptosis (Stegh et al. 2000). Plectin is rich in cysteine residues (34 in humans) half of which are spread over its C-terminal globular domain. One of these, a highly reactive cysteine present in PRD5, has been shown to serve as a target for nitrosylation in vitro and presumably is also the one responsible for plectin’s nitrosylation observed in vivo (Spurny et al. 2007). The phosphorylation site for mitotic CDK1 has been mapped to threonine 4542 (Swiss-Prot: P30427) at the end of PRD 6 (Malecz et al. 1996), and a high-affinity binding site for the ubiquitin ligase Shia E3 to residues 95–117 of plectin isoform 1 (House et al. 2003).

Schematic representation of proteins known to interact directly with plectin. Plectin is shown in the center with the interacting proteins grouped by their cellular location or functional categories. Abbreviations (clockwise from top): HDs hemidesmosomes, TJ tight junctions, FAs focal adhesions, NMJ neuromuscular junction, MFs microfilaments, IFs intermediate filaments, and MTs microtubules. For details see text
Many other interaction partners have been identified, among them the membrane skeleton proteins fodrin and α-spectrin (Brown et al. 2001; Herrmann and Wiche, 1987), desmoplakin (Eger et al. 1997), bullous pemphigoid antigen (BPAG) 1 and 2 (Koster et al. 2003; Steiner-Champliaud et al. 2010), the outer nuclear membrane protein nesprin 3 (Wilhelmsen et al. 2005), the tight junction protein ZO-1 (zona occludens-1; Chen et al. 2006), and skeletal muscle-specific ankyrins (Maiweilidan et al. 2011). The breast cancer susceptibility gene product, BRCA2, which associates with the centrosome during S and early M phase, interacts with plectin forming a complex that controls centrosome localization (Niwa et al. 2009). Furthermore, plectin regulates FUS (fused in sarcoma), an RNA-binding protein involved in transcription, splicing, and mRNA transport, by sequestering it in the cytoplasm (Thomsen et al. 2012).
Lately, it has become clear that plectin, in addition to structurally reinforcing the cytoskeleton, acts as a scaffold for molecules and proteins involved in signaling, by positioning them at specific sites within the cells. Examples are the signaling molecule phosphatidylinositol-4,5-bisphosphate (PIP2; involved in the interaction of plectin with actin; Andrä et al. 1998), the nonreceptor tyrosine kinase Fer (requires plectin to turn down its autophosphorylation; Lunter and Wiche 2002), the receptor for activated C kinase 1 (RACK1; when bound to plectin downregulates PKC signaling during the initial stages of cell adhesion; Osmanagic-Myers and Wiche 2004), the γ-subunit of AMP-activated protein kinase (AMPK; associates with Z-disk-bound plectin in differentiated myofibers; Gregor et al. 2006), components of the MAP kinase Erk 1/2 signaling pathway (required for controlled cell migration; Osmanagic-Myers et al. 2006), the chemokine receptor CXCR4 (plays an important role in stromal-derived factor-1 signaling and trafficking, and in HIV-1 infection; Ding et al. 2008), the RON receptor (a member of the Met proto-oncogen family frequently overexpressed in pancreatic cancer; Yu et al. 2012), and the NR3a subunit of the glutamate receptor (Eriksson et al. 2007).
Plectin and human diseases
In 1996, several groups reported that mutations in the plectin gene (PLEC) were the cause of autosomal recessive forms of the skin disease epidermolysis bullosa simplex associated with muscular dystrophy (EBS-MD; OMIM # 226670). Patients with EBS-MD lack plectin expression in their cells due to premature stop codons coupled with nonsense-mediated mRNA decay. In the meantime, other forms of EBS have been ascribed to mutations in the plectin gene, such as EBS with pyloric atresia (EBS-PA; OMIM #612138) and EBS-MD with myasthenic syndrome (EBS-MD-MyS). The first and thus far only autosomal dominant plectin mutation, known as EBS-Ogna (OMIM #131950), was reported in 2002. Carriers of this mutation suffer from a generalized skin fragility but do not show a muscular phenotype. More recently, also patients, that show only a muscular phenotype without skin abnormalities, have been identified; these patients carry a mutation in the first coding exon of isoform P1f (Gundesli et al. 2010). More details about phenotypes caused by plectin mutations will be given below.
A few recent reports have implicated plectin upregulation in several types of epithelial cancers (Katada et al. 2012; Bausch et al. 2009; Lee et al. 2004; Pawar et al. 2011). Furthermore, plectin has been identified in a phage display screen for biomarkers of pancreatic ductal adenocarcinoma (Kelly et al. 2008). In this type of malignant cells, plectin translocates to the cell membrane, whereas in non-tumor cells, plectin is restricted to the cytoplasmic compartment; concurrently, expression of plectin increases with tumor progression and metastasis (Bausch et al. 2009, Bausch et al. 2011). Due to the specificity of plectin for premalignant and high grade pancreatic lesions, plectin is being developed as an in vivo imaging biomarker for pancreatic cancer (Bausch et al. 2011; Konkalmatt et al. 2013).
Plectin in skin
Expression and subcellular localization
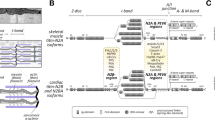
The analysis of plectin transcripts in murine skin revealed expression of all eight plectin isoforms starting with alternative first coding exons (Fuchs et al. 1999). Immunofluorescence microscopy of skin sections showed that P1a expression was restricted to the basal (proliferating) keratinocyte cell layer of interfollicular epidermis, whereas P1c was found in basal as well as suprabasal (differentiating) keratinocyte cell layers (Ackerl et al. 2007; Andrä et al. 2003; Walko et al. 2011) (Fig. 3a). Plectin, presumably P1a, is also expressed in the outer root sheath of hair follicles (Ackerl et al. 2007; Joubeh et al. 2003). Coexpression of P1a and P1c in basal keratinocyte cell cultures has been demonstrated on both, mRNA and protein levels (Andrä et al. 2003; Kostan et al. 2009; Walko et al. 2011), with P1a being expressed at highest level (Andrä et al. 2003). Similar to the in vivo situation, expression of P1c was maintained when cultured keratinocyte monolayers were induced to stratify and undergo terminal differentiation, whereas expression of P1a was down-regulated (Kostan et al. 2009). The most prominently expressed isoforms in epidermal cells other than keratinocytes are P1 (Langerhans cells and Thy 1.2-expressing T lymphocytes; Abrahamsberg et al. 2005) and P1c (melanocytes; unpublished observations of the authors). In dermal fibroblasts, expression of plectin isoforms P1, P1b, P1c, and P1f has been demonstrated (Abrahamsberg et al. 2005; Burgstaller et al. 2010; Winter et al. 2008), and, upon skin wounding, also the macrophages and T lymphocytes infiltrating the dermis are P1 and P1f positive (Abrahamsberg et al. 2005). In addition to full-length plectin isoforms, variants lacking the central rod domain have been detected in cultured keratinocytes and/or dermal fibroblasts on mRNA as well as protein levels (Koster et al. 2004; Natsuga et al. 2010a, b).

Expression of plectin isoforms in mouse skin and schematic models of HDs. a Cryosections of adult mouse skin were subjected to immunofluorescence microscopy using pan-plectin and isoform-specific antibodies as indicated. Asterisks indicate basal cell membrane separating epidermis (top) from dermis (bottom). Note, P1-specific antibodies strongly stained dermal fibroblasts and epidermal dendritic cells, but failed to stain the upper epidermal layers; staining of basal epidermal keratinocytes was negligible. P1a immunoreactivity was restricted to the basal membrane of basal keratinocytes, while P1c-specific signals were more prominent in cells of the upper epidermal layers. Anti-P1f antibodies showed specific but relatively faint peripheral staining of upper layer keratinocytes. Bar 20 μm. b Schematic drawing of the major components of skin HDs. The transmembrane domain proteins integrin α4β6 and BPAG2 bind to laminin 322 (Ln-322), a major component of the extracellular matrix, and to the cytoplasmic proteins plectin and BPAG1, which in turn connect to the keratin IF network. c Hypothetical model for HD stabilization through plectin self-association. P1a clusters integrin α6β4 molecules at the basal cell surface and strengthens the HD through its association with keratin K5/14 filaments generating a vertical force component (arrows pointing upwards). Lateral association of dimeric P1a proteins via their rod domains generates an additional (horizontal) force component that exerts cohesion and compaction (opposing arrows). (b and c are modified from Rezniczek et al. 2010 and Walko et al. 2011, respectively)
Specific functions of isoforms
P1a: key element for assembly and homeostasis of HDs
Based on plectin’s prominence at plasma membrane junctional sites of keratin IFs in the epidermis (Wiche et al. 1983, 1984), it had been anticipated early on that plectin could be a component of HDs and desmosomes. HDs are specialized IF-associated multiprotein complexes that provide stable adhesion of basal epithelial cells to the underlying basement membrane in stratified as well as certain complex and simple epithelia (Margadant et al. 2008), while desmosomal complexes link IFs to sites of intercellular adhesion. In contrast to HDs, desmosome-like structures are also found in non-epithelial tissues, such as heart and skeletal muscle (Delmar and McKenna 2010, Green and Simpson 2007). The high molecular mass IF-associated proteins, HD1 and IFAP300, later identified as orthologs of plectin, had in fact been characterized in early studies as component of HDs and desmosomes in bovine tongue tissue and in a rat epithelial cell line (Hieda et al. 1992; Skalli et al. 1994). The first evidence for isoform P1a being a hemidesmosomal protein was provided by immunogold electron microscopy of rat skin applying isoform-specific antibodies (see Rezniczek et al. 1998, Fig. 9), before a series of subsequent studies clearly established P1a as the plectin isoform specifically associating with HDs (Andrä et al. 2003; Kostan et al. 2009).
Studies in cultured keratinocytes have demonstrated that the association of integrin α6β4 with P1a via the integrin β4 subunit and their clustering is crucial for the formation of HDs. This initial P1a/integrin β4 association fosters the recruitment of BPAG2 and BPAG1e molecules to the complex and the anchorage of the keratin IF network through P1a and BPAG1e follows (Hopkinson and Jones 2000; Koster et al. 2003; Litjens et al. 2006). Additional stabilization of HDs against mechanical stress appears to be mediated by lateral interaction of P1a molecules via their rod domains (Walko et al. 2011). This notion is also supported by the fact that rodless isoforms can only partially substitute for full-length P1a in maintaining HD function in skin of EBS-MD patients (Koster et al. 2004; Natsuga et al. 2010a, b). Lateral aggregation of P1a molecules in HDs also appears to protect P1a from degradation via calpains, a family of calcium-dependent proteases that could be involved in HD destruction to mediate migration and terminal differentiation (Kostan et al. 2009; Walko et al. 2011). The importance of the integrin subunit β4-P1a interaction is confirmed in vivo by the much reduced level or complete absence of HDs in mice deficient in either plectin, or integrin subunits α6 and β4, which are born with large patches of detached epidermis and generally die within a few hours after birth (Andrä et al. 1997; Dowling et al. 1996; Georges-Labouesse et al. 1996; van der Neut et al. 1996). Similarly, the anchorage of keratin IFs at HDs was found to be compromised in mice carrying null alleles of either plectin or BPAG1e (Andrä et al. 1997; Guo et al. 1995), or in knock-in mice carrying the dominant negative Ogna mutation (Walko et al. 2011). Furthermore, underscoring the importance of P1a for HD function, skin blistering due to compromised assembly and function of HDs was observed in EBS-Ogna mice (having reduced P1a levels), but not in mice that were deficient in P1c (Fuchs et al. 2009; Walko et al. 2011).
Studies of adult mice carrying conditional deletions of either plectin or the integrin subunit α6 in the epidermis indicated that in vivo HDs are very stable structures, exhibiting a very low turnover (Ackerl et al. 2007; Niculescu et al. 2011). However, under certain circumstances, such as wound healing or carcinoma invasion, HDs in migrating and proliferating keratinocytes turn into highly dynamic structures, similar to keratinocytes cultivated ex vivo where HDs assemble and disassemble continuously (Geuijen and Sonnenberg 2002; Ozawa et al. 2010). The mechanism of HD disassembly has been addressed in several studies. It was shown that P1a binds to integrin subunit β4 via multiple interaction domains including its N-terminal ABD (de Pereda et al. 2009; Koster et al. 2004; Rezniczek et al. 1998). Growth factor-induced elevations of intracellular Ca2+ levels lead to the activation of PKCα that phosphorylates the integrin β4 subunit on serine and threonine residues located in domains mediating the interaction with plectin (Frijns et al. 2012; Margadant et al. 2008; Rabinovitz et al. 2004), while Ca2+/calmodulin interacts with P1a’s ABD (Kostan et al. 2009). Both events cause the dissociation of P1a from membrane-bound integrin followed by a redistribution of both proteins and disassembly of HDs (see Kostan et al. 2009, Fig. 9).
P1c: regulator of MT dynamics in keratinocytes
The immunolocalization of P1c, the second major isoform of plectin expressed in basal keratinocytes, reveals partial colocalization with MTs, mostly in a dotted pattern (Fig. 4a). Unexpectedly, and contrary to the function of other cytolinker proteins previously analyzed, P1c has recently been shown to act as a MT destabilizer (Valencia et al. 2013). Compared to wild-type cells, MTs in P1c-deficient keratinocytes were found to be more resistant toward nocodazole-induced disassembly and to display increased acetylation of tubulin, a modification indicating reduced dynamic behavior. In addition, live imaging of MTs in P1c-deficient as well as in plectin-null cells revealed decreased MT dynamics. Accordingly, increased MT stability in keratinocytes due to P1c deficiency led to changes in a number of cellular properties and functions known to be MT dependent, including altered cell shape, increased velocity but loss of directionality of migration, smaller-sized focal adhesions, higher glucose uptake due to increased transfer of glucose transporters to the plasma membrane, and mitotic spindle aberrations combined with reduced growth rates of cells.

P1c: a MT destabilizer. a Deconvolved immunofluorescence microscopy images of cytoplasmic regions of wild-type (left and middle panel) and P1c-deficient (right panel) keratinocytes. Antibodies used are indicated; merged triple- or double-channel images are shown. Note dotted decoration of MTs by both, P1c and MAP2, in wild-type cells, whereas in P1c-deficient cells, MTs were decorated with MAPs over much longer distances. b Proposed mechanism of P1c-mediated MT destabilization. P1c, anchored to IFs via its C-terminal IF-docking site (red sphere), binds to MTs via its isoform-specific N-terminal sequence (including the ABD), while the SH3 domain located within the spectrin repeats (SH3/SR) binds to MAPs. Interference with the MT-stabilizing function of MAPs ensures a dynamic MT network in wild-type cells (left panel), whereas in P1c-deficient cells, more MAPs can bind along MTs leading to their stabilization (right panel). (a and b correspond to parts of Fig. 9 in Valencia et al. 2013)
A mechanism proposed for microtubule destabilization through P1c, based on ex vivo and in vitro assays, is depicted by the cartoon shown in Fig. 4b. According to this model, P1c is targeted to MTs via its isoform-specific N-terminal sequence (including the ABD), while one of its further downstream molecular domains, the SH3-like domain embedded within its spectrin repeats (plakin domain), interacts with MT-associated proteins (MAPs), known stabilizers of MT polymers. Since the binding of plectin’s SH3 domain antagonizes the MT-stabilizing and assembly promoting function of MAPs, P1c interaction with MTs leads to MT destabilization, which in turn favors a dynamic state of the MT network in cells. On the other hand, when P1c is absent, such as in P1c-deficient cells, more MAPs are bound along MTs (Fig. 4a) leading to the stabilization of the polymer (Fig. 4b). Based on experiments in which truncated forms of P1c were expressed into P1c-deficient keratinocytes to assess their potential for rescuing MT deficits, it was concluded that normal MT properties could be restored in P1c-deficient keratinocytes only if P1c variants were used that contained the C-terminal IF-binding domain of the molecule and thus were able to associate with the keratin IF network (Valencia et al. 2013). Hence, beyond revealing an unexpected and novel function of P1c and plectin in general, this study opened an intriguing new perspective on how the cytoskeleton regulates its dynamics by uncovering the potential of IFs to destabilize MTs via an associated cytolinker protein, thereby stimulating MT dynamics.
P1: the missing link to the nucleus of skin cells and a role in migration?
Plectin has been shown to bind to the outer nuclear envelope protein nesprin 3α, thereby linking IFs to the nucleus in keratinocytes, fibroblasts, and other cell types (Ketema et al. 2007; Wilhelmsen et al. 2005). It remains to be established which plectin isoform is associated with nesprin 3α. P1 would definitively be a good candidate, as studies in muscle tissue indicated that P1 is a major linker component between IFs and the nuclear/ER membrane (Konieczny et al. 2008; Rezniczek et al. 2007). Indeed, P1 transcripts have been identified in cultured keratinocytes (Andrä et al. 2003).
The analysis of primary dermal fibroblasts derived from isoform P1-deficient mice revealed abnormalities in the actin cytoskeleton and an impaired migration potential of the cells (Abrahamsberg et al. 2005), reminiscent of plectin-null fibroblasts (Andrä et al. 1998). Along these lines, in skin wound healing experiments carried out with P1-deficient mice, reduced leukocyte infiltration was observed during in vivo wound healing, and P1-deficient T cells isolated from lymph nodes of these mice showed diminished chemotactic migration ex vivo (Abrahamsberg et al. 2005). While these findings demonstrate that P1 is involved in the regulation of leukocyte recruitment during skin wound healing, the underlying mechanism remains to be determined. Nuclear positioning via P1-IF networking could possibly play a role.
Special functions of other isoforms expressed in epidermal and/or dermal skin cells, particularly of P1f, have not been analyzed. Based on P1f’s predominantly peripheral localization in keratinocytes (Fig. 3a), fibroblasts, and myofibers (see below), a role of this isoform in actin-network regulation (Andrä et al. 1998), in addition to its IF-anchoring function, is not unexpected.
Human skin disease phenotypes caused by plectin mutations
In 1996, several groups reported that patients suffering from the skin blistering disease epidermolysis bullosa simplex with muscular dystrophy, lacked plectin expression in skin and muscle tissues due to defects in the plectin gene (Chavanas et al. 1996; Gache et al. 1996; McLean et al. 1996; Pulkkinen et al. 1996; Smith et al. 1996). Since then, mutations within the plectin gene were identified in an increasing number of EBS variants with skin involvement. These variants include EBS-MD, EBS-MD-MyS, EBS-PA, and EBS-Ogna (reviewed in Rezniczek et al. 2010; Winter and Wiche 2013). In all plectin-associated EBS variants, skin blistering is caused by trauma-induced rupture of basal epidermal keratinocytes in their cytoplasm, usually at a level just above the basal cell membrane-anchored HDs. Cell integrity in the suprabasal keratinocyte cell layers is generally not affected, and cell–cell interactions are preserved.
Until now, there are reports of 30 independent cases of EBS-MD, in which the diagnosis was confirmed by identifying the mutation (see Table 1 in Winter and Wiche 2013; and additional cases reported in Charlesworth et al. 2013). At the molecular level, EBS-MD is frequently associated with nonsense, insertion, or deletion mutations, resulting in premature stop codons within exon 31, the exon that encodes plectin’s coiled coil rod domain. This predicts truncated polypeptides and mRNA downregulation through nonsense-mediated mRNA decay. As processing of the rodless variant of plectin remains unaltered by mutations in exon 31 and as the expression of the rodless isoform has been detected in cells of EBS-MD patients (Koster et al. 2004; Natsuga et al. 2010a, b), it has been postulated that rodless plectin could partially rescue some basic functions of plectin in association with its N- and C-terminal domains. EBS-MD was further found to be caused by several in-frame insertion and deletion mutations in exons located up- or downstream of exon 31 (Charlesworth et al. 2013; Winter and Wiche 2013). It is likely that these mutations affect specific protein–protein interactions relevant for the proper functioning of plectin in HDs. In general, clinical phenotypes of affected individuals vary considerably with respect to severity of skin blistering. In addition to congenital skin blistering, deformation of the nails and tooth decay were observed in a number of EBS-MD patients, and in some cases, erosive lesions on the oral or laryngeal mucosa, hoarseness, respiratory complications during infancy, and urethral strictures have been reported (Babic et al. 2010; Charlesworth et al. 2013; Kunz et al. 2000; Schara et al. 2004).
EBS with pyloric atresia (EBS-PA) is one of the most severe EBS variants. It manifests as severe generalized neonatal skin blistering and gastric abnormalities (pyloric or duodenal atresia) and frequently leads to early postnatal demise of affected individuals. Eleven patients with EBS-PA have been reported so far carrying homozygous or compound heterozygous mutations (Charlesworth et al. 2013; Winter and Wiche 2013). The majority (9) of EBS-PA-associated mutations have been localized in exons encoding the N-terminal region of plectin, some (6) were found in exon 32 encoding the C-terminal domain, and only one (situated at the very distal portion of exon 31) affects the rod domain of plectin. This specific localization pattern of mutations suggests that the EBS-PA phenotype could result from genetic mutations specifically affecting the N- and C-terminal portions of plectin that are harboring the integrin β4- and IF-binding sites. Moreover, since these mutations are mostly outside of the rod, premature termination will lead to loss of expression of both, full-length and rodless, plectin isoforms (Charlesworth et al. 2003; Natsuga et al. 2010a), and thus to a more severe phenotype. Interestingly, a first case of non-lethal EBS-PA improving with age has recently been reported (Charlesworth et al. 2013). This clinical phenotype was found to be caused by two compound heterozygous mutations resulting in premature termination codons within exons 30 and 32. Considering that one of the mutations allows the synthesis of a truncated version of the plectin molecule missing only its most C-terminal part (PRD 6 and tail) and that low but detectable expression of plectin has been demonstrated in the patient’s epidermis, it has been proposed that the truncated protein, which still harbors the critical residues for integrin α6β4- and IF-binding, could partially compensate for the loss of plectin expression from the other allele (Charlesworth et al. 2013).
EBS-MD-MyS is a rare EBS variant that has been diagnosed in three patients with compound heterozygous PLEC mutations (Banwell et al. 1999; Forrest et al. 2010; Selcen et al. 2011), and additionally in one patient with a homozygous insertion mutation in the gene encoding the acetylcholine receptor subunit CHRNE (Maselli et al. 2010). Three of the patients revealed marked skin blistering from birth, whereas one exhibited only sparse signs of epidermolysis bullosa (Banwell et al. 1999; Forrest et al. 2010; Selcen et al. 2011).
Besides the recessive forms of plectin-linked EBS, there is one autosomal dominant form designated EBS-Ogna. This rare mutation was originally identified in a kindred near the small Norwegian town of Ogna (hence its name) and later in four unrelated German families and two Dutch individuals (Bolling 2010a; Gedde-Dahl 1971; Kiritsi et al. 2013; Koss-Harnes et al. 2002). EBS-Ogna is caused by a heterozygous missense mutation, R2000 W, within plectin’s rod-encoding exon 31 (Kiritsi et al. 2013; Koss-Harnes et al. 2002). In contrast to the other more severe plectin-associated EBS variants, EBS-Ogna has a mild course with localized acral skin blistering and formation of erosions, which predominate with increasing age (Kiritsi et al. 2013; Koss-Harnes et al. 2002). Other skin-related symptoms encompass posttraumatic violaceous macules, hypopigmented macules, mild focal palmoplantar keratoderma, and pitting of the teeth (Kiritsi et al. 2013; Koss-Harnes et al. 2002). EBS-Ogna patients do not develop MD, nor do they show signs of PA. The molecular mechanisms underlying the unique skin phenotype have been shown to involve the isoform-specific proteolytic degradation of P1a in HDs of basal keratinocytes (Walko et al. 2011). Besides being the only mutation with an autosomal dominant type of inheritance reported thus far, the Ogna mutation is emerging as the most frequent mutation of plectin described in families of different ethnic origins, suggesting that it may represent a hotspot for mutations within the plectin gene.
Mouse models for plectin-related skin blistering diseases
To mimic the clinical features of plectin-associated skin blistering diseases in an animal model, several transgenic mouse lines have been generated. The first was a plectin-null mouse (Andrä et al. 1997), followed by a conditional knockout mouse line, where the ablation of plectin was restricted to keratin 5-expressing epithelia (K5-Cre/cKO; Ackerl et al. 2007). Within 1 to 2 days after birth (occasionally even at birth), plectin-null mice begin to show signs of gross skin blistering, especially at fore- and hindlimbs and, in some cases, around the mouth and nasal cavities (Andrä et al. 1997). In K5-Cre/cKO mice, skin blistering develops with a similar onset, body distribution, and severity (Ackerl et al. 2007). In some cases, plectin-null and K5-Cre/cKO mice are born with denuded paws (authors’ unpublished observations), resembling aplasia cutis congenita, similar to the most severe cases reported for plectin-associated EBS-PA (Charlesworth et al. 2003, 2013; Nakamura et al. 2005; Pfendner and Uitto 2005). In plectin-null mice, blisters on the lower extremities are often hemorrhagic (Andrä et al. 1997), which, however, is not observed in K5-Cre/cKO mice (authors’ unpublished observations). This type of bleeding might indicate a possible perturbation of vascular endothelial cells, which normally express plectin at relatively high levels (Errante et al. 1994; Wiche et al. 1983). Epidermal desmosomes and consequently cell–cell interactions were found to be unaffected by keratinocyte-specific deletion of plectin (Ackerl et al. 2007). Inspection of the upper gastrointestinal tract of newborn K5-Cre/cKO mice after at least one nursing revealed the presence of multiple blisters on their palates and in some cases also on their tongues but not in the esophagus. Thus, plectin knockout mice seem to die from malnutrition as a consequence of blistering of the oral epithelia and possibly obstruction of the oral cavity by especially large blisters or cellular debris (Ackerl et al. 2007). Recently, a mouse line carrying the EBS-Ogna mutation has been generated (Walko et al. 2011). EBS-Ogna mice mimic the human disease by displaying mild skin fragility but no extracutaneous pathologies. The skin phenotype of EBS-Ogna mice reflects sparse and dysfunctional HDs, and insufficient protein levels of (HD-associated) P1a, whereas P1c is maintained at normal levels (Walko et al. 2011). Noteworthy, HD deficiency in basal keratinocytes of the epidermis could be partially restored by treating mice with calpain inhibitors, setting the stage for further exploration of treatment strategies for the disease.
Plectin in skeletal muscle
Expression and subcellular localization
The original immunolocalization of plectin in mammalian tissues and isolated cardiomyocytes showed a prominent association with desmin at Z-disks of skeletal muscle, dense plaques of smooth muscle, and Z-disks and intercalated disks of cardiac muscle (Wiche et al. 1983; Zernig and Wiche 1985). Using immunogold labeling, a number of studies extended these findings to the ultrastructural level. It was shown that plectin-positive filamentous structures radiating from Z-disks were linked to desmin IFs generating lateral linkages among adjacent Z-disks (Hijikata et al. 1999, Reipert et al. 1999; Schröder et al. 1999) and that similar bridges were connecting Z-disks of peripheral myofibrils to sarcolemmal densities presumed to represent costameric remnants (Hijikata et al. 2003; Reipert et al. 1999; Rezniczek et al. 2007; Schröder et al. 1999). Furthermore, plectin was shown to be associated with desmin IFs linking myofibrils to mitochondria at the level of the Z-disk and along the entire length of the sarcomere (Reipert et al. 1999). These studies strongly suggested that the desmin IF network in partnership with plectin was forming an extensively connected three-dimensional intermyofibrillar and subplasma membrane protein scaffold that physically linked the contractile apparatus to internal organelles and to junctional complexes at the sarcolemma, thus providing a mechanical continuum between internal myofiber structures and the extracellular matrix surrounding the fiber.
Specific functions of isoforms
In skeletal muscle tissue, all eight of the originally identified transcript variants containing alternatively spliced first coding exons were found to be expressed, although for some of the first exons, very weak signals were obtained by RNase protection assays (Fuchs et al. 1999). Moreover, a tissue-specific dominance of isoform P1d was detected, which was most prominent in skeletal muscle and heart. Beyond P1d, the muscle-specific set of major isoforms comprises P1, P1b, and P1f, with relative transcript levels of >10:4:3:1 for P1d, P1f, P1b, and P1, respectively (Fuchs et al. 1999). Visualization of GFP fusion proteins upon forced expression in cells (Rezniczek et al. 2003, 2007), and immunofluorescence microscopy of teased muscle fibers using isoform-specific antibodies (Konieczny et al. 2008), demonstrated that the various isoforms were targeted to distinct subcellular locations, where, as it turned out, they are fulfilling distinct functions (Fig. 5a; see also Fig. 8 in Konieczny et al. 2008).

Plectin in skeletal muscle. a Schematic representation of plectin’s isoform diversity and specific functions. The four major isoforms expressed in muscle, P1d (yellow), P1f (dark blue), P1b (green), and P1 (purple) are site-specifically bound via their N-terminal molecular domains (not indicated) to Z-disks (P1d), the costameric/transmembrane dystrophin-glycoprotein-complex (DGC) (P1f), mitochondrial organelles (P1b), and the nuclear/ER membrane system (P1). The various isoforms recruit (via their C-terminal IF-binding domain) the desmin IF network (blue lines) to their specific locations, thereby creating a densely interwoven resilient filamentous network that mechanically integrates the contractile apparatus with cytoplasmic organelles, the sarcolemma, and the extracellular compartment. b Immunofluorescence microscopy of teased muscle fibers from wild-type and conditional muscle-restricted knockout (MCK-Cre/cKO) mice. Visualized proteins are indicated; myonuclei are stained in blue. Note massive formation of desmin-positive protein aggregates and misaligned and disoriented Z-disks, both hallmarks of myofibrillar myopathies, in specimens of mutant mice (corresponds to Fig. 2D in Konieczny et al. 2008)
P1d: the Z-disk anchor protein of desmin filaments
P1d is the only isoform of plectin that is specifically expressed in skeletal and cardiac muscle, but not in other tissues (Fuchs et al. 1999). When GFP fusion proteins of P1d were expressed in differentiated myoblasts, they were found to be targeted exclusively to Z-disks (Rezniczek et al. 2007). Furthermore, when cryosections of skeletal muscle from isoform-specific P1d knockout mice were immunolabeled using antibodies recognizing all isoforms (anti-pan-plectin), only the sarcolemma and dotty remnants in the interior of fibers were stained, contrasting the highly ordered cross-striated staining pattern typical of wild-type tissue. Similarly, in fibers of P1d-deficient muscle, plectin signals were preserved in peripheral, perinuclear, and costameric regions, where P1 and P1f were still present. Moreover, in P1d-deficient muscle tissue and teased muscle fibers, aggregates of desmin IFs were observed in the interior but not peripheral areas of the fiber. While these observations clearly show that P1d is responsible for the recruitment and anchorage of desmin IFs to Z-disks, the question how P1d itself is specifically targeted to Z-disk structures is still unsolved. It will be a challenging task to answer this question considering that the isoform-specific sequence of P1d preceding its ABD (common to all isoforms) comprises only 5 amino acid residues.
P1f: plugging the contractile apparatus to the sarcolemma
In fibroblasts, P1f was shown to be an integral component of mature, focal adhesion-evolved fibrillar adhesions, which are characterized by their robust, fibronectin fibril-aligned, elongated structure, and central subcellular localization. P1f-mediated vimentin IF network formation at these sites creates a resilient cage-like core structure of IFs that encases and positions the nucleus while being stably connected to the exterior of the cell (Burgstaller et al. 2010). The situation in muscle fibers seems to be similar. The forced expression of GFP-labeled P1f in differentiated myotubes showed that the fusion protein was targeting exclusively to the sarcolemma where it associated with the dystrophin-glycoprotein complex (DGC; Rezniczek et al. 2007). In fact, plectin binds directly to dystrophin and the transmembrane laminin receptor β-dystroglycan, two of the major DGC constituent proteins, via two independent binding sites located in its N-terminal portion (see Fig. 5), leaving its C-terminal IF-binding site freely accessible (Rezniczek et al. 2007). In this way, P1f can act as a well-anchored docking site for desmin IF networks that link costameres (and associated structures in the extracellular compartment) with peripheral myofibrils, mitochondria, and myonuclei (see Fig. 5a). Based, among others, on the strikingly similar expression patterns of P1f and dystrophin during differentiation of myoblasts to myotubes, a role of P1f in the formation and maturation of costameres has been suggested (Rezniczek et al. 2007), reminiscent of the transformation of focal adhesions to fibrillary adhesion in fibroblasts. In certain types of mature muscle fibers, such as fast type 2A fibers of quadriceps muscle from adult mice, P1f could additionally be found at Z-disks, suggesting a certain degree of interchangeability of plectin isoforms once IF-docking sites have been formed and become functional (Rezniczek et al. 2007).
What seems to be most crucial for myofiber integrity is the structural axis generated by P1d and P1f that interlinks sarcomeres at their Z-disks and connects them with the plasma membrane. Disruption of one (P1d) or both (P1f and P1d) of these IF-linking elements inevitably leads to the loss of muscle fiber integrity. In fact, as demonstrated by isoform-specific gene targeting, elimination of P1d alone leads to the formation of aggregates in the interior of the fiber, but not in its peripheral sarcolemmal regions. Despite that in this case, the remaining P1f was able to preserve costameric IF structures; isoform P1d knockout mice showed a similarly severe muscle phenotype as the conditional knockout mice (lacking all isoforms), or desmin-null mice (Konieczny et al. 2008).
P1b: integrating mitochondria into the desmin IF network
Colocalizing with mitochondria, P1b acts as a direct linker between the organelle and the desmin network. This linkage has been demonstrated for IFs of the vimentin and desmin type, but most likely occurs also with other types of IFs. The exon 1b-encoded sequence of this isoform serves as a mitochondrial targeting and anchoring signal, inserting the protein into the outer mitochondrial membrane via its N-terminal part. The bulk of the protein remains in the cytosol, where it interacts via its C-terminal high-affinity binding site with IFs recruiting them to the organelle (Winter et al. 2008). In fact, the loss of P1b leads to marked shape changes of mitochondrial networks, manifesting as elongation of mitochondrial networks in P1b-deficient fibroblasts and myoblasts (Winter et al. 2008). Moreover, in muscle tissue that is deficient in P1d (but not P1b), association of residual plectin with mitochondria and colocalization of collapsed desmin IFs with mitochondrial aggregates were observed; whereas in plectin-null muscle, similar to P1b-deficient muscle, this was not the case (Konieczny et al. 2008). The underlying mechanism of mitochondrial network deregulation through P1b deficiency and its biological significance remain to be determined. However, there is evidence that mitochondrion-bound plectin, in addition to having a positioning effect, serves as a scaffolding platform for signaling complexes (Winter et al. 2008).
P1: linking desmin IFs to the nuclear/ER membrane system
In muscle fibers, contrary to other isoforms, P1 localizes primarily to the outer nuclear/ER membrane system (Rezniczek et al. 2007, Konieczny et al. 2008). Exon 1 encodes a putative nuclear localization signal (NLS), and N-terminal fragments of P1 up to a certain length exhibit nuclear localization (Rezniczek et al. 2003). Thus, the NLS domain is likely to play a role in recruiting P1 to the nuclear/ER membrane, where upon binding to other local interaction partners, it forms a recruitment platform for IFs. One of these putative proteins has been identified as nesprin-3, a KASH domain-containing protein that interacts with plectin’s ABD (Ketema et al. 2007). As nesprin-3 is an outer nuclear membrane protein, it provides via plectin a direct link between the nuclear envelope and the cytoplasmic IF network (Ketema and Sonnenberg 2011). Other nuclear/ER membrane-associated proteins that bind directly either to the isoform-specific sequence or to other molecular domains of P1 are expected to be found. In addition to the nuclear/ER membrane system, P1 showed partial association with the sarcolemma in certain types of fully differentiated muscle fibers (Rezniczek et al. 2007). Thus, like P1f, P1 may show spatial interchangeability with other isoforms at mature or late stages of myofiber development.
The isoform-specific N-terminal sequence of P1 (180 amino acid residues) is the longest of all known plectin isoforms. One of its specific features is that it contains a binding site of unusual high activity for the substrate-binding site of Siah, a ring domain protein functioning in E3 ubiquitin ligase complexes that mediate degradation of a wide range of cellular proteins. It has therefore been suggested that P1 could serve as a dominant negative regulator of Siah proteins (House et al. 2003).
Skeletal muscle phenotypes of plectinopathy patients
Plectin has been found to be prominently expressed in muscle cells early on, but its importance for the structure and function of muscle became clearly evident only when patients with defects in the plectin gene were found to suffer, in addition to skin blistering, from late onset muscular dystrophy (for a recent review see Winter and Wiche 2013). The clinical phenotypes of patients vary considerably with respect to onset and progression of muscular symptoms as well as severity of skin blistering. Skeletal muscle weakness usually starts in the second decade of life and shows relatively slow disease progression leading to a loss of ambulation after 10–20 years by affecting facial, extraocular, limb, and trunk muscles. Moreover, patients with cardiac pathology have been reported that display asymptomatic left ventricular hypertrophy (Schröder et al. 2002) and adult-onset biventricular dilated cardiomyopathy (Bolling et al. 2010b). Similar to skin, immunofluorescence microscopy of skeletal muscle tissue of EBS-MD patients revealed either drastically reduced or completely absent plectin-specific staining (McMillan et al. 2007). While antibodies recognizing epitopes in the central rod domain of plectin in general lack immunoreactivity, antibodies directed against plectin’s C- or N-terminal regions often revealed specific signals, indicating the expression of rodless or truncated plectin versions, which might attenuate the clinical phenotypes (Koster et al. 2004; Natsuga et al. 2010a, b). Immunolabeling for desmin usually reveals the formation of cytoplasmic and subsarcolemmal desmin-positive protein aggregates. Moreover, ultrastructural analyses of skeletal muscles showed disarrayed myofibrils and loss of sarcomeric organization, accompanied by pathological changes of membranous organelles and neuromuscular endplates (Bauer et al. 2001; McMillan et al. 2007; Schröder et al. 2002). Therefore, most plectinopathies can be assigned to the group of myofibrillar myopathies (MFMs), an expanding group of clinically and genetically heterogeneous muscle diseases characterized by misfolded proteins and pathological desmin-positive protein aggregates, myofibrillar degeneration, and mitochondrial abnormalities (Ferrer and Olive 2008; Schröder and Schoser 2009). EBA-PA cases do not display signs of muscular pathology; however, onset of MD is to be expected if patients survive long enough (Charlesworth et al. 2013, Winter and Wiche 2013).
Muscular symptoms are displayed also by patients suffering from EBS-MD-MyS or LGMD2Q. EBS-MD-MyS patients show first signs of muscle weakness accompanied by myasthenic symptoms within the first decade of life. In fact, defects in neuromuscular transmission, as demonstrated by a pathological decremental response in neuromuscular transmission studies, were observed in all cases. Recently, a homozygous 9-bp deletion in the isoform-specific sequence of P1f was found to cause limb-girdle muscular dystrophy type Q2 (LGMD2Q), characterized by a muscular pathology without any skin involvement (Gundesli et al. 2010). In this case, the probands suffered from early-onset limb-girdle syndrome with loss of ambulation in their late twenties. As distinct isoforms perform different functions, mutations in other isoforms can be expected to be identified in the future and should be considered in cases of undiagnosed muscular dystrophies.
Mouse models for plectin-related muscular dystrophies
Plectin-null mice, similar to plectinopathy patients, show abnormalities reminiscent of minicore myopathies manifesting in skeletal muscle as focal loss of myofibrils and disruption of sarcomeres affecting Z-lines and adjacent myofibrils, Z-band streaming, and increased amounts of necrotic fibers (Andrä et al. 1997), and in heart as disintegration of intercalated disks. However, as these mice die within 2–3 days after birth, they are of limited use as animal models for plectin-related muscular dystrophies. To overcome this problem, a conditional, striated muscle-restricted plectin knockout mouse line (MCK-Cre/cKO) was generated. Closely mimicking the human disease, these mice show progressive degenerative alterations, including aggregation and partial loss of desmin IFs, aggregation and dysfunction of mitochondria, and a partial detachment of the contractile apparatus from the sarcolemma (Konieczny et al. 2008). The analysis of these mice showed that the disruption of myofibrils, and particular of Z-disks, most likely occurs as a consequence of disruptive contractions of myofibrils. Due to the loss of its anchorage via plectin and the resulting defective lateral alignment of myofibrils, the desmin IF network can no longer keep sarcomeres in register. The additional formation of dysfunctional protein (desmin) aggregates in subplasma membrane and interior cellular compartments contributes to the deleterious phenotypes of plectin-deficient myofibers. Mutant mice, mimicking the complex disease pattern of human plectinopathies, and ex vivo myocyte cell cultures derived from them, thus provide powerful tools for elucidating functional and molecular consequences of plectin gene defects leading to skeletal muscle pathology.
For three of the four major plectin isoforms expressed in muscle, P1d, P1b, and P1, isoform-specific knockout mouse lines were generated, which lack just one of these isoforms, while expressing all others. Some of their phenotypes have been described above. In addition, a double knockout mouse line, deficient in P1d and desmin has been generated (Konieczny et al. 2008), and a muscle-restricted conditional plectin knockout line lacking dystrophin in addition to plectin has recently become available (Raith et al. 2013). The phenotypic analysis of the latter unveiled a new facet of plectin’s ever increasing functional repertoire by establishing a mechanistic link between sarcolemma-associated plectin, its microtubule-destabilizing action, and glucose transporter–mediated glucose uptake of muscle fibers (Raith et al. 2013).
Plectin in Schwann cells of peripheral nerve
Similar to skin and muscle tissues, plectin shows widespread occurrence in neural cells and tissues (Errante et al. 1994), and neurodegeneration has been reported in some cases of EBS-MD (Smith et al. 1996, Bauer et al. 2001). Plectin has also been implied in Alexander disease, a neurological disorder caused by mutations in GFAP (Tian et al. 2006). The phenotypic analysis of isoform P1c-specific knockout mice revealed slightly reduced neurofilament densities in some of their axons and significantly reduced nerve conduction velocity in sciatic nerve (Fuchs et al. 2009). Whether these alterations are mechanistically linked remains to be shown. With respect to the diversity and specific functions of isoforms, the best studied neural cell system are myelinating Schwann cells of the peripheral nerve.
Expression and isoform diversity
Immunolabeling of murine sciatic nerve using isoform-specific antibodies revealed expression of P1 and P1c in the cytoplasm and at the periphery of axons as well as in the cytoplasm of myelinating Schwann cells (Fig. 6a; see also Fuchs et al. 2009; Walko et al. 2013). In addition, relatively strong P1-specific labeling of some neurofilament-negative cells, probably representing endoneural fibroblasts was noticed. P1a, the predominantly in stratified epithelial tissues–expressed isoform, could not be detected in axons or myelinating Schwann cells, but was found to be expressed in putative endoneural fibroblasts; in these cells, also P1f, one of the major isoforms of dermal fibroblasts (Burgstaller et al. 2010) was detected. Faint, dotted signals of P1f were also observed in axons, whereas myelinating Schwann cells were found to be P1f negative. When the expression of isoforms in cultures of primary Schwann cells and nerve fibroblasts was assessed by immunoblotting of cell lysates, the expression of full-length versions of isoforms P1, P1c, and P1f was demonstrated for both types of cells. In addition, in Schwann cell lysates, the corresponding rodless isoform variants lacking the central rod domain were detected at relatively high levels, whereas in nerve fibroblast samples, a rodless version was found only in the case of P1 (Walko et al., 2013). Consistent with its in situ visualization in tissues (Fig. 6a), P1a was detectable in lysates of nerve fibroblast, but not Schwann cell cultures (Walko et al. 2013).

Plectin in peripheral nerve. a Frozen sciatic nerve cross sections were immunolabeled using antibodies specific to P1c, P1, P1a, and P1f as indicated; sections were double labeled using antibodies to neurofilament (NF) M chain to specifically label axons; nuclei were labeled with Hoechst dye (blue). Arrowheads, myelinating Schwann cells; arrows, neurofilament-negative/plectin-positive cells. Bar 10 μm. b and c Association of plectin with vimentin IFs in Schwann cells. Teased sciatic nerve fibers (b) or cultured primary Schwann cells (c) were double immunolabeled using antibodies to plectin (not discriminating between isoforms) and vimentin. Bars 20 μm (b), 10 μm (c). Note codistribution of plectin and vimentin in Cajal bands and transverse trabeculae (arrowheads in b), and in radial and axial lamellipodial protrusions (arrows in c, left and right panel, respectively). Also note colocalization of plectin with short filamentous vimentin IF intermediates (arrowheads). (Reprinted in modified form from Walko et al. 2013)
Subcellular distribution
Confocal immunofluorescence microscopy of teased sciatic nerve fibers using antibodies that do not discriminate between isoforms revealed codistribution of plectin and vimentin IFs in longitudinal Cajal bands and transversing trabeculae (Fig. 6b; Walko et al. 2013) which constitute the cytoplasm of myelinating Schwann cells (Court et al. 2004). In cultured Schwann cells, plectin was found to colocalize with the vimentin IF network as well as with vimentin-positive short filamentous structures (Fig. 6c) that most likely were representing filament precursors, as previously reported for fibroblast cells (Spurny et al. 2008). Coimmunoprecipitation and immunolocalization experiments revealed that plectin present in Cajal bands formed a tight complex not only with vimentin but also with β-dystroglycan, the core component of the DGC (Walko et al. 2013). Thus, by anchoring vimentin IFs to dystroglycan, plectin fulfills in myelinating Schwann cells a similar function as in skeletal muscle where it anchors the desmin network to the DGC (Rezniczek et al. 2007).
Specific functions of isoforms
As recently shown, the binding of plectin to β-dystroglycan and the ensuing recruitment of the vimentin IF network to the abaxonal Schwann cell membrane contribute to the stabilization of the dystroglycan complex (Walko et al. 2013). The Schwann cell-specific ablation of plectin in a protein zero (P0)-Cre/conditional plectin knockout mouse line (P0-Cre/cKO) was found to lead to faulty vimentin IF-dystroglycan complex formation, leading to an aggravation of aging-related destabilization of the myelin sheath (Walko et al. 2013). In contrast to skeletal muscle, where the loss of plectin causes massive aggregation of desmin IFs (Konieczny et al. 2008), in myelinating Schwann cells, the impact of plectin deficiency on the organization of the vimentin IF system was detectable just as a decrease in the vimentin IF network density in a fraction of nerve fibers characterized by large caliber axons (Walko et al. 2013). One possible explanation for this difference could be that peripheral nerves in general are more protected and thus less exposed to mechanical stress compared to skeletal muscle fibers. The isoform of plectin acting as the specific interaction partner of β-dystroglycan in differentiating Schwann cells has still to be identified. Given the absence of P1f in this type of cells (see Fig. 6a), the strongest candidate for this function is P1, which has previously been shown to interact with the DGC component α-dystrobrevin-1 (Hijikata et al. 2008). In this context, it is noteworthy that albeit the P1a-specific binding partner integrin β4 is expressed in myelinating Schwann cells (Nodari et al. 2008), cell-extracellular matrix junctions similar to HDs are not formed by this type of cell, at least not at its fully differentiated state, due to the lack of isoform P1a expression.
Concerning potential roles of other Schwann cell-expressed plectin isoforms, it is of interest that P1f is abundantly expressed in ex vivo cultures of Schwann cells, in contrast to fully differentiated (myelinating) tissue cells. Given the established role of this isoform in regulating focal adhesion turnover and cell shape in fibroblasts (Burgstaller et al. 2010), its expression in ex vivo cell cultures would be consistent with a role in cell migration, a feature presumably required by precursor and regenerating, but not fully differentiated Schwann cells. The role of P1c, the other major plectin isoform identified in Schwann cells, presently is unknown. Whether it performs MT-related functions in Schwann cells, as it does in keratinocytes, remains to be tested. Even if no alterations in the organization of MTs were observed in Cajal bands of sciatic nerves from myelinating Schwann cell-specific knockout mice (Walko et al. 2013), P1c could still play a role in the regulation of MT-dependent transport processes in the cytoplasm of these cells. Of note, in contrast to isoform P1c knockout mice, a defect in motor nerve conduction velocity of sciatic nerve could not be detected in conditional, Schwann cell-specific plectin knockout (P0-Cre/cKO) mice (authors’unpublished data), suggesting that this phenotype is specifically related to axonal P1c.
Perspective
Plectin has emerged as a preeminent cytoskeletal linker protein controlling IF functions. Studies on skin, skeletal muscle fiber, and Schwann cells, the systems discussed in this review article, clearly showed that distinct cell types express specific sets of plectin isoforms that are targeted to distinct cellular locations. By recruiting IF networks to their target sites, plectin isoforms control interior cytoarchitecture as well as cell shape, with in part dramatic effects on mechanical resilience as well as an array of other basic cellular function, among them proliferation, metabolism, migration, and signal transduction. In light of plectin’s isoform diversity and IF partnership in practically all mammalian cell types, the challenge for future relevant research lays in the analysis of isoform-specific functions and their mechanisms in systems other than the highly stress-prone tissues discussed above. It will be of particular interest to unravel the roles of plectin isoforms in the development and function of the brain and of motor neurons in the peripheral nervous system, as for both cases, there is evidence for important physiological roles of plectin, based on phenotypic analyses of plectinopathy patients as well as plectin knockout mice; and a similar situation pertains to the vascular system.
References
Abrahamsberg C, Fuchs P, Osmanagic-Myers S, Fischer I, Propst F, Elbe-Bürger A, Wiche G (2005) Targeted ablation of plectin isoform 1 uncovers role of cytolinker proteins in leukocyte recruitment. Proc Natl Acad Sci USA 102(51):18449–18454
Ackerl R, Walko G, Fuchs P, Fischer I, Schmuth M, Wiche G (2007) Conditional targeting of plectin in prenatal and adult mouse stratified epithelia causes keratinocyte fragility and lesional epidermal barrier defects. J Cell Sci 120:2435–2443
Andrä K, Lassmann H, Bittner R, Shorny S, Fässler R, Propst F, Wiche G (1997) Targeted inactivation of plectin reveals essential function in maintaining the integrity of skin, muscle, and heart cytoarchitecture. Genes Dev 11:3143–3156
Andrä K, Nikolic B, Stöcher M, Drenckhahn D, Wiche G (1998) Not just scaffolding: plectin regulates actin dynamics in cultured cells. Genes Dev 12:3442–3451
Andrä K, Kornacker I, Jörgl A, Zörer M, Spazierer D, Fuchs P, Fischer I, Wiche G (2003) Plectin-isoform-specific rescue of hemidesmosomal defects in plectin (−/−) keratinocytes. J Invest Dermatol 120:189–197
Babic I, Karaman-Ilic M, Pustisek N, Susic S, Skaric I, Kljenak A, Cikojevic D (2010) Respiratory tract involvement in a child with epidermolysis bullosa simplex with plectin deficiency: a case report. Int J Pediatr Otorhinolaryngol 74:302–305
Banwell BL, Russel J, Fukudome T, Shen XM, Stilling G, Engel AG (1999) Myopathy, myasthenic syndrome, and epidermolysis bullosa simplex due to plectin deficiency. J Neuropathol Exp Neurol 58:832–846
Bauer JW, Rouan F, Kofler B, Rezniczek GA, Kornacker I, Muss W, Hametner R, Klausegger A, Huber A, Pohla-Gubo G et al (2001) A compound heterozygous one amino-acid insertion/nonsense mutation in the plectin gene causes epidermolysis bullosa simplex with plectin deficiency. Am J Pathol 158:617–625
Bausch D, Mino-Kenudson M, Fernández-Del Castillo C, Warshaw AL, Kelly KA, Thayer SP (2009) Plectin-1 is a biomarker of malignant pancreatic intraductal papillary mucinous neoplasms. J Gastrointest Surg 13:1948–1954
Bausch D, Thomas S, Mino-Kenudson M, Fernández-del CC, Bauer TW, Williams M, Warshaw AL, Thayer SP, Kelly KA (2011) Plectin-1 as a novel biomarker for pancreatic cancer. Clin Cancer Res 17:302–309
Bhattacharya R, Gonzalez AM, Debiase PJ, Trejo HE, Goldman RD, Flitney FW, Jones JC (2009) Recruitment of vimentin to the cell surface by beta3 integrin and plectin mediates adhesion strength. J Cell Sci 122:1390–1400
Bolling MC (2010a) Epidermolysis bullosa simplex New insights in desmosomal cardiocutaneous syndromes. http://irs.ub.rug.nl/ppn/326355561 Dissertation. University of Groningen, The Netherlands
Bolling MC, Pas HH, de Visser M, Aronica E, Pfendner EG, van den Berg MP, Diercks GF, Suurmeijer AJ, Jonkman MF (2010b) PLEC1 mutations underlie adult-onset dilated cardiomyopathy in epidermolysis bullosa simplex with muscular dystrophy. J Invest Dermatol 130:1178–1181
Brown MJ, Hallam JA, Liu Y, Yamada KM, Shaw S (2001) Cutting edge: integration of human T lymphocyte cytoskeleton by the cytolinker plectin. J Immunol 167:641–645
Burgstaller G, Gregor M, Winter L, Wiche G (2010) Keeping the vimentin network under control: cell-matrix adhesion-associated plectin 1f affects cell shape and polarity of fibroblasts. Mol Biol Cell 21:3362–3375
Charlesworth A, Gagnoux-Palacios L, Bonduelle M, Ortonne JP, De Raeve L, Meneguzzi G (2003) Identification of a lethal form of epidermolysis bullosa simplex associated with a homozygous genetic mutation in plectin. J Invest Dermatol 121:1344–1348
Charlesworth A, Chiaverini C, Chevrant-Breton J, Delrio M, Diociaiuti A, Dupuis RP, El-Achem M, Le Fiblec B, Sankari-Ho AM, Valhquist A, Wierzbicka E, Lacour JP, Meneguzzi G (2013) Epidermolysis bullosa simplex with PLEC mutations: new phenotypes and new mutations. Br J Dermatol 168:808–814
Chavanas S, Pulkkinen L, Gache Y, Smith FJ, McLean WH, Uitto J, Ortonne JP, Meneguzzi G (1996) A homozygous nonsense mutation in the PLEC1 gene in patients with epidermolysis bullosa simplex with muscular dystrophy. J Clin Invest 98:2196–2200
Chen VC, Li X, Perreault H, Nagy JI (2006) Interaction of zonula occludens-1 (ZO-1) with alpha-actinin-4: application of functional proteomics for identification of PDZ domain-associated proteins. J Proteome Res 5:2123–2134
Clubb BH, Chou YH, Herrmann H, Svitkina TM, Borisy GG, Goldman RD (2000) The 300-kDa intermediate filament-associated protein (IFAP300) is a hamster plectin ortholog. Biochem Biophys Res Commun 273:183–187
Court FA, Sherman DL, Pratt T, Garry EM, Ribchester RR, Cottrell DF, Fleetwood-Walker SM, Brophy PJ (2004) Restricted growth of Schwann cells lacking Cajal bands slows conduction in myelinated nerves. Nature 431:191–195
de Pereda JM, Lillo MP, Sonnenberg A (2009) Structural basis of the interaction between integrin alpha6beta4 and plectin at the hemidesmosomes. EMBO J 28:1180–1190
Delmar M, McKenna WJ (2010) The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res 107:700–714
Ding Y, Zhang L, Goodwin JS, Wang Z, Liu B, Zhang J, Fan GH (2008) Plectin regulates the signaling and trafficking of the HIV-1 co-receptor CXCR4 and plays a role in HIV-1 infection. Exp Cell Res 314:590–602
Dowling J, Yu QC, Fuchs E (1996) Beta4 integrin is required for hemidesmosome formation, cell adhesion and cell survival. J Cell Biol 134:559–572
Eger A, Stockinger A, Wiche G, Foisner R (1997) Polarisation-dependent association of plectin with desmoplakin and the lateral submembrane skeleton in MDCK cells. J Cell Sci 110:1307–1316
Elliott CE, Becker B, Oehler S, Castañón MJ, Hauptmann R, Wiche G (1997) Plectin transcript diversity: identification and tissue distribution of variants with distinct first coding exons and rodless isoforms. Genomics 42:115–125
Eriksson M, Nilsson A, Samuelsson H, Samuelsson EB, Mo L, Akesson E, Benedikz E, Sundström E (2007) On the role of NR3A in human NMDA receptors. Physiol Behav 92:54–59
Errante LD, Wiche G, Shaw G (1994) Distribution of plectin, an intermediate filament-associated protein, in the adult rat central nervous system. J Neurosci Res 37:515–528
Favre B, Schneider Y, Lingasamy P, Bouameur JE, Begré N, Gontier Y, Steiner-Champliaud MF, Frias MA, Borradori L, Fontao L (2011) Plectin interacts with the rod domain of type III intermediate filament proteins desmin and vimentin. Eur J Cell Biol 90:390–400
Ferrer I, Olive M (2008) Molecular pathology of myofibrillar myopathies. Expert Rev Mol Med 10:e25
Foisner R, Wiche G (1987) Structure and hydrodynamic properties of plectin molecules. J Mol Biol 198:515–531
Foisner R, Leichtfried FE, Herrmann H, Small JV, Lawson D, Wiche G (1988) Cytoskeleton-associated plectin: in situ localization, in vitro reconstitution, and binding to immobilized intermediate filament proteins. J Cell Biol 106:723–733
Foisner R, Traub P, Wiche G (1991) Protein kinase A- and protein kinase C-regulated interaction of plectin with lamin B and vimentin. Proc Natl Acad Sci USA 88:3812–3816
Foisner R, Bohn W, Mannweiler K, Wiche G (1995) Distribution and ultrastructure of plectin arrays in subclones of rat glioma C6 cells differing in intermediate filament protein (vimentin) expression. J Struct Biol 115:304–317
Foisner R, Malecz N, Dressel N, Stadler C, Wiche G (1996) M-phase-specific phosphorylation and structural rearrangement of the cytoplasmic cross-linking protein plectin involve p34cdc2 kinase. Mol Biol Cell 7:273–288
Fontao L, Geerts D, Kuikman I, Koster J, Kramer D, Sonnenberg A (2001) The interaction of plectin with actin: evidence for cross-linking of actin filaments by dimerization of the actin-binding domain of plectin. J Cell Sci 114:2065–2076
Forrest K, Mellerio JE, Robb S, Dopping-Hepenstal PJ, McGrath JA, Liu L, Buk SJ, Al-Sarraj S, Wraige E, Jungbluth H (2010) Congenital muscular dystrophy, myasthenic symptoms and epidermolysis bullosa simplex (EBS) associated with mutations in the PLEC1 gene encoding plectin. Neuromuscul Disord 20:709–711
Frijns E, Kuikman I, Litjens S, Raspe M, Jalink K, Ports M, Wilhelmsen K, Sonnenberg A (2012) Phosphorylation of threonine 1736 in the C-terminal tail of integrin beta4 contributes to hemidesmosome disassembly. Mol Biol Cell 23:1475–1485
Fuchs P, Zörer M, Rezniczek GA, Spazierer D, Oehler S, Castañón MJ, Hauptmann R, Wiche G (1999) Unusual 5’ transcript complexity of plectin isoforms: novel tissue-specific exons modulate actin binding activity. Hum Mol Genet 8:2461–2472
Fuchs P, Spazierer D, Wiche G (2005) Plectin rodless isoform expression and its detection in mouse brain. Cell Mol Neurobiol 25:1141–1150
Fuchs P, Zörer M, Reipert S, Rezniczek GA, Propst F, Walko G, Fischer I, Bauer J, Leschnik MW, Lüscher B, Thalhammer JG, Lassmann H, Wiche G (2009) Targeted inactivation of a developmentally regulated neural plectin isoform (plectin 1c) in mice leads to reduced motor nerve conduction velocity. J Biol Chem 284:26502–26509. doi:M109.018150
Gache Y, Chavanas S, Lacour JP, Wiche G, Owaribe K, Meneguzzi G, Ortonne JP (1996) Defective expression of plectin/HD1 in epidermolysis bullosa simplex with muscular dystrophy. J Clin Invest 97:2289–2298
Gache V, Waridel P, Winter C, Juhem A, Schroeder M, Shevchenko A, Popov AV (2010) Xenopus meiotic microtubule-associated interactome. PLoS ONE 5:e9248
García-Alvarez B, Bobkov A, Sonnenberg A, de Pereda JM (2003) Structural and functional analysis of the actin binding domain of plectin suggests alternative mechanisms for binding to F-actin and integrin beta4. Structure 11:615–625
Gedde-Dahl T Jr (1971) Epidermolysis Bullosa: a clinical, genetic and epidemiological study. Johns Hopkins University Press, Baltimore
Geerts D, Fontao L, Nievers MG, Schaapveld RQ, Purkis PE, Wheeler GN, Lane EB, Leigh IM, Sonnenberg A (1999) Binding of integrin alpha6beta4 to plectin prevents plectin association with F-actin but does not interfere with intermediate filament binding. J Cell Biol 147:417–434
Georges-Labouesse E, Messaddeq N, Yehia G, Cadalbert L, Dierich A, Le Meur M (1996) Absence of integrin alpha 6 leads to epidermolysis bullosa and neonatal death in mice. Nat Genet 13:370–373
Geuijen CA, Sonnenberg A (2002) Dynamics of the alpha6beta4 integrin in keratinocytes. Mol Biol Cell 13:3845–3858
Green KJ, Simpson CL (2007) Desmosomes: new perspectives on a classic. J Invest Dermatol 127:2499–2510
Gregor M, Zeöld A, Oehler S, Marobela KA, Fuchs P, Weigel G, Hardie DG, Wiche G (2006) Plectin scaffolds recruit energy-controlling AMP-activated protein kinase (AMPK) in differentiated myofibres. J Cell Sci 119:1864–1875
Gundesli H, Talim B, Korkusuz P, Balci-Hayta B, Cirak S, Akarsu NA, Topaloglu H, Dincer P (2010) Mutation in exon 1f of PLEC, leading to disruption of plectin isoform 1f, causes autosomal-recessive limb-girdle muscular dystrophy. Am J Hum Genet 87:834–841
Guo L, Degenstein L, Dowling J, Yu QC, Wollmann R, Perman B, Fuchs E (1995) Gene targeting of BPAG1: abnormalities in mechanical strength and cell migration in stratified epithelia and neurologic degeneration. Cell 81:233–243
Herrmann H, Wiche G (1983) Specific in situ phosphorylation of plectin in detergent-resistant cytoskeletons from cultured Chinese hamster ovary cells. J Biol Chem 258:14610–14618
Herrmann H, Wiche G (1987) Plectin and IFAP-300 K are homologous proteins binding to microtubule-associated proteins 1 and 2 and to the 240-kilodalton subunit of spectrin. J Biol Chem 262:1320–1325
Hieda Y, Nishizawa Y, Uematsu J, Owaribe K (1992) Identification of a new hemidesmosomal protein, HD1: a major, high molecular mass component of isolated hemidesmosomes. J Cell Biol 116:1497–1506
Hijikata T, Murakami T, Imamura M, Fujimaki N, Ishikawa H (1999) Plectin is a linker of intermediate filaments to Z-discs in skeletal muscle fibers. J Cell Sci 112:867–876
Hijikata T, Murakami T, Ishikawa H, Yorifuji H (2003) Plectin tethers desmin intermediate filaments onto subsarcolemmal dense plaques containing dystrophin and vinculin. Histochem Cell Biol 119:109–123
Hijikata T, Nakamura A, Isokawa K, Imamura M, Yuasa K, Ishikawa R, Kohama K, Takeda S, Yorifuji H (2008) Plectin 1 links intermediate filaments to costameric sarcolemma through beta-synemin, alpha-dystrobrevin and actin. J Cell Sci 121:2062–2074
Hopkinson SB, Jones JC (2000) The N terminus of the transmembrane protein BP180 interacts with the N-terminal domain of BP230, thereby mediating keratin cytoskeleton anchorage to the cell surface at the site of the hemidesmosome. Mol Biol Cell 11:277–286
House CM, Frew IJ, Huang HL, Wiche G, Traficante N, Nice E, Catimel B, Bowtell DD (2003) A binding motif for Siah ubiquitin ligase. Proc Natl Acad Sci USA 100:3101–3106
Inoue T, Pecen P, Maddala R, Skiba NP, Pattabiraman PP, Epstein DL, Rao PV (2010) Characterization of cytoskeleton-enriched protein fraction of the trabecular meshwork and ciliary muscle cells. Invest Ophthalmol Vis Sci 51:6461–6471
Janda L, Damborský J, Rezniczek GA, Wiche G (2001) Plectin repeats and modules: strategic cysteines and their presumed impact on cytolinker functions. BioEssays 23:1064–1069
Jarrett HW, Foster JL (1995) Alternate binding of actin and calmodulin to multiple sites on dystrophin. J Biol Chem 270:5578–5586
Joubeh S, Mori O, Owaribe K, Hashimoto T (2003) Immunofluorescence analysis of the basement membrane zone components in human anagen hair follicles. Exp Dermatol 12:365–370
Katada K, Tomonaga T, Satoh M, Matsushita K, Tonoike Y, Kodera Y, Hanazawa T, Nomura F, Okamoto Y (2012) Plectin promotes migration and invasion of cancer cells and is a novel prognostic marker for head and neck squamous cell carcinoma. J Proteomics 75:1803–1815
Kelly KA, Bardeesy N, Anbazhagan R, Gurumurthy S, Berger J, Alencar H, Depinho RA, Mahmood U, Weissleder R (2008) Targeted nanoparticles for imaging incipient pancreatic ductal adenocarcinoma. PLoS Med 5:e85
Ketema M, Sonnenberg A (2011) Nesprin-3: a versatile connector between the nucleus and the cytoskeleton. Biochem Soc Trans 39:1719–1724
Ketema M, Wilhelmsen K, Kuikman I, Janssen H, Hodzic D, Sonnenberg A (2007) Requirements for the localization of nesprin-3 at the nuclear envelope and its interaction with plectin. J Cell Sci 120:3384–3394
Kiritsi D, Pigors M, Tantcheva-Poor I, Wessel C, Arin MJ, Kohlhase J, Bruckner-Tuderman L, Has C (2013) Epidermolysis bullosa simplex ogna revisited. J Invest Dermatol 133:270–273
Konieczny P, Fuchs P, Reipert S, Kunz WS, Zeöld A, Fischer I, Paulin D, Schröder R, Wiche G (2008) Myofiber integrity depends on desmin network targeting to Z-disks and costameres via distinct plectin isoforms. J Cell Biol 181:667–681
Konkalmatt PR, Deng D, Thomas S, Wu MT, Logsdon CD, French BA, Kelly KA (2013) Plectin-1 targeted AAV vector for the molecular imaging of pancreatic cancer. Front Oncol 3:84
Koss-Harnes D, Hoyheim B, Anton-Lamprecht I, Gjesti A, Jorgensen RS, Jahnsen FL, Olaisen B, Wiche G, Gedde-Dahl T Jr (2002) A site-specific plectin mutation causes dominant epidermolysis bullosa simplex Ogna: two identical de novo mutations. J Invest Dermatol 118:87–93
Kostan J, Gregor M, Walko G, Wiche G (2009) Plectin isoform-dependent regulation of keratin-integrin alpha6beta4 anchorage via Ca2+/calmodulin. J Biol Chem 284:18525–18536
Koster J, Geerts D, Favre B, Borradori L, Sonnenberg A (2003) Analysis of the interactions between BP180, BP230, plectin and the integrin alpha6beta4 important for hemidesmosome assembly. J Cell Sci 116:387–399
Koster J, van Wilpe S, Kuikman I, Litjens SH, Sonnenberg A (2004) Role of binding of plectin to the integrin beta4 subunit in the assembly of hemidesmosomes. Mol Biol Cell 15:1211–1223
Koszka C, Leichtfried FE, Wiche G (1985) Identification and spatial arrangement of high molecular weight proteins (Mr 300 000–330 000) co-assembling with microtubules from a cultured cell line (rat glioma C6). Eur J Cell Biol 38:149–156
Kunz M, Rouan F, Pulkkinen L, Hamm H, Jeschke R, Bruckner-Tuderman L, Brocker EB, Wiche G, Uitto J, Zillikens D (2000) Mutation reports: epidermolysis bullosa simplex associated with severe mucous membrane involvement and novel mutations in the plectin gene. J Invest Dermatol 114:376–380
Lee KY, Liu YH, Ho CC, Pei RJ, Yeh KT, Cheng CC, Lai YS (2004) An early evaluation of malignant tendency with plectin expression in human colorectal adenoma and adenocarcinoma. J Med 35:141–149
Lie AA, Schröder R, Blümcke I, Magin TM, Wiestler OD, Elger CE (1998) Plectin in the human central nervous system: predominant expression at pia/glia and endothelia/glia interfaces. Acta Neuropathol 96:215–221
Lieska N, Yang HY, Goldman RD (1985) Purification of the 300 K intermediate filament-associated protein and its in vitro recombination with intermediate filaments. J Cell Biol 101:802–813
Litjens SH, Koster J, Kuikman I, van Wilpe S, de Pereda JM, Sonnenberg A (2003) Specificity of binding of the plectin actin-binding domain to beta4 integrin. Mol Biol Cell 14:4039–4050
Litjens SH, de Pereda JM, Sonnenberg A (2006) Current insights into the formation and breakdown of hemidesmosomes. Trends Cell Biol 16:376–383
Liu CG, Maercker C, Castañon MJ, Hauptmann R, Wiche G (1996) Human plectin: organization of the gene, sequence analysis, and chromosome localization (8q24). Proc Natl Acad Sci USA 93:4278–4283
Lo SH, Janmey PA, Hartwig JH, Chen LB (1994) Interactions of tensin with actin and identification of its three distinct actin-binding domains. J Cell Biol 125:1067–1075
Lunter PC, Wiche G (2002) Direct binding of plectin to Fer kinase and negative regulation of its catalytic activity. Biochem Biophys Res Commun 296:904–910
Mack GJ, Compton DA (2001) Analysis of mitotic microtubule-associated proteins using mass spectrometry identifies astrin, a spindle-associated protein. Proc Natl Acad Sci USA 98:14434–14439
Maiweilidan Y, Klauza I, Kordeli E (2011) Novel interactions of ankyrins-G at the costameres: the muscle-specific Obscurin/Titin-Binding-related Domain (OTBD) binds plectin and filamin C. Exp Cell Res 317:724–736
Malecz N, Foisner R, Stadler C, Wiche G (1996) Identification of plectin as a substrate of p34cdc2 kinase and mapping of a single phosphorylation site. J Biol Chem 271:8203–8208
Margadant C, Frijns E, Wilhelmsen K, Sonnenberg A (2008) Regulation of hemidesmosome disassembly by growth factor receptors. Curr Opin Cell Biol 20:589–596
Maselli R, Arredondo J, Cagney O, Mozaffar T, Skinner S, Yousif S, Davis R, Gregg J, Sivak M, Konia T, Thomas K, Wollmann R (2010) Congenital myasthenic syndrome associated with epidermolysis bullosa caused by homozygous mutations in PLEC1 and CHRNE. Clin Genet 80:444–451
McInroy L, Määttä A (2011) Plectin regulates invasiveness of SW480 colon carcinoma cells and is targeted to podosome-like adhesions in an isoform-specific manner. Exp Cell Res 317:2468–2478
McLean WH, Pulkkinen L, Smith FJ, Rugg EL, Lane EB, Bullrich F, Burgeson RE, Amano S, Hudson DL, Owaribe K, McGrath JA, McMillan JR, Eady RA, Leigh IM, Christiano AM, Uitto J (1996) Loss of plectin causes epidermolysis bullosa with muscular dystrophy: cDNA cloning and genomic organization. Genes Dev 10:1724–1735
McMillan JR, Akiyama M, Rouan F, Mellerio JE, Lane EB, Leigh IM, Owaribe K, Wiche G, Fujii N, Uitto J et al (2007) Plectin defects in epidermolysis bullosa simplex with muscular dystrophy. Muscle Nerve 35:24–35
Nakamura H, Sawamura D, Goto M, McMillan JR, Park S, Kono S, Hasegawa S, Paku S, Nakamura T, Ogiso Y, Shimizu H (2005) Epidermolysis bullosa simplex associated with pyloric atresia is a novel clinical subtype caused by mutations in the plectin gene (PLEC1). J Mol Diagn 7:28–35
Natsuga K, Nishie W, Akiyama M, Nakamura H, Shinkuma S, McMillan JR, Nagasaki A, Has C, Ouchi T, Ishiko A, Hirako Y, Owaribe K, Sawamura D, Bruckner-Tuderman L, Shimizu H (2010a) Plectin expression patterns determine two distinct subtypes of epidermolysis bullosa simplex. Hum Mutat 31:308–316
Natsuga K, Nishie W, Shinkuma S, Arita K, Nakamura H, Ohyama M, Osaka H, Kambara T, Hirako Y, Shimizu H (2010b) Plectin deficiency leads to both muscular dystrophy and pyloric atresia in epidermolysis bullosa simplex. Hum Mutat 31:1687–1698
Niculescu C, Ganguli-Indra G, Pfister V, Dupe V, Messaddeq N, De Arcangelis A, Georges-Labouesse E (2011) Conditional ablation of integrin alpha-6 in mouse epidermis leads to skin fragility and inflammation. Eur J Cell Biol 90:270–277
Nikolic B, Mac Nulty E, Mir B, Wiche G (1996) Basic amino acid residue cluster within nuclear targeting sequence motif is essential for cytoplasmic plectin-vimentin network junctions. J Cell Biol 134:1455–1467
Niwa T, Saito H, Imajoh-ohmi S, Kaminishi M, Seto Y, Miki Y, Nakanishi A (2009) BRCA2 interacts with the cytoskeletal linker protein plectin to form a complex controlling centrosome localization. Cancer Sci 100:2115–2125
Nodari A, Previtali SC, Dati G, Occhi S, Court FA, Colombelli C, Zambroni D, Dina G, Del Carro U, Campbell KP, Quattrini A, Wrabetz L, Feltri ML (2008) Alpha6beta4 integrin and dystroglycan cooperate to stabilize the myelin sheath. J Neurosci 28:6714–6719
Okumura M, Uematsu J, Hirako Y, Nishizawa Y, Shimizu H, Kido N, Owaribe K (1999) Identification of the hemidesmosomal 500 kDa protein (HD1) as plectin. J Biochem 126:1144–1150
Osmanagic-Myers S, Wiche G (2004) Plectin-RACK1 (receptor for activated C kinase 1) scaffolding: a novel mechanism to regulate protein kinase C activity. J Biol Chem 279:18701–18710
Osmanagic-Myers S, Gregor M, Walko G, Burgstaller G, Reipert S, Wiche G (2006) Plectin-controlled keratin cytoarchitecture affects MAP kinases involved in cellular stress response and migration. J Cell Biol 174:557–568
Ozawa T, Tsuruta D, Jones JC, Ishii M, Ikeda K, Harada T, Aoyama Y, Kawada A, Kobayashi H (2010) Dynamic relationship of focal contacts and hemidesmosome protein complexes in live cells. J Invest Dermatol 130:1624–1635
Pawar H, Kashyap MK, Sahasrabuddhe NA, Renuse S, Harsha HC, Kumar P, Sharma J, Kandasamy K et al (2011) Quantitative tissue proteomics of esophageal squamous cell carcinoma for novel biomarker discovery. Cancer Biol Ther 12:510–522
Pfendner E, Uitto J (2005) Plectin gene mutations can cause epidermolysis bullosa with pyloric atresia. J Invest Dermatol 124:111–115
Pulkkinen L, Smith FJ, Shimizu H, Murata S, Yaoita H, Hachisuka H, Nishikawa T, McLean WH, Uitto J (1996) Homozygous deletion mutations in the plectin gene (PLEC1) in patients with epidermolysis bullosa simplex associated with late-onset muscular dystrophy. Hum Mol Genet 5:1539–1546
Pytela R, Wiche G (1980) High molecular weight polypeptides (270,000–340,000) from cultured cells are related to hog brain microtubule-associated proteins but copurify with intermediate filaments. Proc Natl Acad Sci USA 77:4808–4812
Rabinovitz I, Tsomo L, Mercurio AM (2004) Protein kinase C-alpha phosphorylation of specific serines in the connecting segment of the beta 4 integrin regulates the dynamics of type II hemidesmosomes. Mol Cell Biol 24:4351–4360
Raith M, Valencia RG, Fischer I, Orthofer M, Penninger JM, Spuler S, Rezniczek GA, Wiche G (2013) Linking cytoarchitecture to metabolism: sarcolemma-associated plectin affects glucose uptake by destabilizing microtubule networks in mdx myofibers. Skeletal muscle (in press)
Reipert S, Steinböck F, Fischer I, Bittner RE, Zeöld A, Wiche G (1999) Association of mitochondria with plectin and desmin intermediate filaments in striated muscle. Exp Cell Res 252:479–491
Rezniczek GA, de Pereda JM, Reipert S, Wiche G (1998) Linking integrin alpha6beta4-based cell adhesion to the intermediate filament cytoskeleton: direct interaction between the beta4 subunit and plectin at multiple molecular sites. J Cell Biol 141:209–225
Rezniczek GA, Abrahamsberg C, Fuchs P, Spazierer D, Wiche G (2003) Plectin 5′-transcript diversity: short alternative sequences determine stability of gene products, initiation of translation and subcellular localization of isoforms. Hum Mol Genet 12:3181–3194
Rezniczek GA, Konieczny P, Nikolic B, Reipert S, Schneller D, Abrahamsberg C, Davies KE, Winder SJ, Wiche G (2007) Plectin 1f scaffolding at the sarcolemma of dystrophic (mdx) muscle fibers through multiple interactions with beta-dystroglycan. J Cell Biol 176:965–977
Rezniczek GA, Walko G, Wiche G (2010) Plectin gene defects lead to various forms of epidermolysis bullosa simplex. Dermatol Clin 28:33–41
Sauer G, Körner R, Hanisch A, Ries A, Nigg EA, Silljé HH (2005) Proteome analysis of the human mitotic spindle. Mol Cell Proteomics 4:35–43
Schara U, Tucke J, Mortier W, Nusslein T, Rouan F, Pfendner E, Zillikens D, Bruckner-Tuderman L, Uitto J, Wiche G, Schroder R (2004) Severe mucous membrane involvement in epidermolysis bullosa simplex with muscular dystrophy due to a novel plectin gene mutation. Eur J Pediatr 163:218–222
Schröder R, Schoser B (2009) Myofibrillar myopathies: a clinical and myopathological guide. Brain Pathol 19:483–492
Schröder R, Warlo I, Herrmann H, van der Ven PF, Klasen C, Blumcke I, Mundegar RR, Fürst DO, Goebel HH, Magin TM (1999) Immunogold EM reveals a close association of plectin and the desmin cytoskeleton in human skeletal muscle. Eur J Cell Biol 78:288–295
Schröder R, Kunz WS, Rouan F, Pfendner E, Tolksdorf K, Kappes-Horn K, Altenschmidt-Mehring M, Knoblich R, van der Ven PF, Reimann J et al (2002) Disorganization of the desmin cytoskeleton and mitochondrial dysfunction in plectin-related epidermolysis bullosa simplex with muscular dystrophy. J Neuropathol Exp Neurol 61:520–530
Selcen D, Juel VC, Hobson-Webb LD, Smith EC, Stickler DE, Bite AV, Ohno K, Engel AG (2011) Myasthenic syndrome caused by plectinopathy. Neurology 76:327–336
Sevcík J, Urbániková L, Kostan J, Janda L, Wiche G (2004) Actin-binding domain of mouse plectin. Crystal structure and binding to vimentin. Eur J Biochem 271:1873–1884
Skalli O, Jones JC, Gagescu R, Goldman RD (1994) IFAP 300 is common to desmosomes and hemidesmosomes and is a possible linker of intermediate filaments to these junctions. J Cell Biol 125:159–170
Smith FJ, Eady RA, Leigh IM, McMillan JR, Rugg EL, Kelsell DP, Bryant SP, Spurr NK, Geddes JF, Kirtschig G, Milana G, de Bono AG, Owaribe K, Wiche G, Pulkkinen L, Uitto J, McLean WH, Lane EB (1996) Plectin deficiency results in muscular dystrophy with epidermolysis bullosa. Nat Genet 13:450–457
Sonnenberg A, Liem RK (2007) Plakins in development and disease. Exp Cell Res 313:2189–2203
Sonnenberg A, Rojas AM, de Pereda JM (2007) The structure of a tandem pair of spectrin repeats of plectin reveals a modular organization of the plakin domain. J Mol Biol 368:1379–1391
Spurny R, Abdoulrahman K, Janda L, Rünzler D, Köhler G, Castañón MJ, Wiche G (2007) Oxidation and nitrosylation of cysteines proximal to the intermediate filament (IF)-binding site of plectin: effects on structure and vimentin binding and involvement in IF collapse. J Biol Chem 282:8175–8187
Spurny R, Gregor M, Castañón MJ, Wiche G (2008) Plectin deficiency affects precursor formation and dynamics of vimentin networks. Exp Cell Res 314:3570–3580
Stegh AH, Herrmann H, Lampel S, Weisenberger D, Andrä K, Seper M, Wiche G, Krammer PH, Peter ME (2000) Identification of the cytolinker plectin as a major early in vivo substrate for caspase 8 during CD95- and tumor necrosis factor receptor-mediated apoptosis. Mol Cell Biol 20:5665–5679
Steinböck FA, Nikolic B, Coulombe PA, Fuchs E, Traub P, Wiche G (2000) Dose-dependent linkage, assembly inhibition and disassembly of vimentin and cytokeratin 5/14 filaments through plectin’s intermediate filament-binding domain. J Cell Sci 113:483–491
Steiner-Champliaud MF, Schneider Y, Favre B, Paulhe F, Praetzel-Wunder S, Faulkner G, Konieczny P, Raith M, Wiche G, Adebola A, Liem RK, Langbein L, Sonnenberg A, Fontao L, Borradori L (2010) BPAG1 isoform-b: complex distribution pattern in striated and heart muscle and association with plectin and alpha-actinin. Exp Cell Res 316:297–313
Svitkina TM, Verkhovsky AB, Borisy GG (1996) Plectin sidearms mediate interaction of intermediate filaments with microtubules and other components of the cytoskeleton. J Cell Biol 135:991–1007
Thomsen C, Udhane S, Runnberg R, Wiche G, Ståhlberg A, Aman P (2012) Fused in sarcoma (FUS) interacts with the cytolinker protein plectin: implications for FUS subcellular localization and function. Exp Cell Res 318:653–661
Tian R, Gregor M, Wiche G, Goldman JE (2006) Plectin regulates the organization of glial fibrillary acidic protein in Alexander disease. Am J Pathol 168:888–897
Valencia RG, Walko G, Janda L, Novacek J, Mihailovska E, Reipert S, Andrä-Marobela K, Wiche G (2013) Intermediate filament-associated cytolinker plectin 1c destabilizes microtubules in keratinocytes. Mol Biol Cell 24:768–784
van der Neut R, Krimpenfort P, Calafat J, Niessen CM, Sonnenberg A (1996) Epithelial detachment due to absence of hemidesmosomes in integrin beta 4 null mice. Nat Genet 13:366–369
Walko G, Vukasinovic N, Gross K, Fischer I, Sibitz S, Fuchs P, Reipert S, Jungwirth U, Berger W, Salzer U, Carugo O, Castañón MJ, Wiche G (2011) Targeted proteolysis of plectin Isoform 1a accounts for hemidesmosome dysfunction in mice mimicking the dominant skin blistering disease EBS-Ogna. PLoS Genet 7(12):e1002396
Walko G, Wögenstein K, Winter L, Fischer I, Feltri L, Wiche G (2013) Stabilization of the dystroglycan complex in Cajal bands of myelinating Schwann cells through plectin-mediated anchorage of vimentin filaments. Glia (in press)
Wiche G, Winter L (2011) Plectin isoforms as organizers of intermediate filament cytoarchitecture. Bioarchitecture 1:14–20
Wiche G, Krepler R, Artlieb U, Pytela R, Denk H (1983) Occurrence and immunolocalization of plectin in tissues. J Cell Biol 97:887–901
Wiche G, Krepler R, Artlieb U, Pytela R, Aberer W (1984) Identification of plectin in different human cell types and immunolocalization at epithelial basal cell surface membranes. Exp Cell Res 155:43–49
Wiche G, Becker B, Luber K, Weitzer G, Castañón MJ, Hauptmann R, Stratowa C, Stewart M (1991) Cloning and sequencing of rat plectin indicates a 466-kD polypeptide chain with a three-domain structure based on a central alpha-helical coiled coil. J Cell Biol 114:83–99
Wilhelmsen K, Litjens SH, Kuikman I, Tshimbalanga N, Janssen H, van den Bout I, Raymond K, Sonnenberg A (2005) Nesprin-3, a novel outer nuclear membrane protein, associates with the cytoskeletal linker protein plectin. J Cell Biol 171:799–810
Winter L, Wiche G (2013) The many faces of plectin and plectinopathies: pathology and mechanisms. Acta Neuropathol 125:77–93
Winter L, Abrahamsberg C, Wiche G (2008) Plectin isoform 1b mediates mitochondrion-intermediate filament network linkage and controls organelle shape. J Cell Biol 181:903–911
Yang Y, Dowling J, Yu QC, Kouklis P, Cleveland DW, Fuchs E (1996) An essential cytoskeletal linker protein connecting actin microfilaments to intermediate filaments. Cell 86:655–665
Yaoita E, Wiche G, Yamamoto T, Kawasaki K, Kihara I (1996) Perinuclear distribution of plectin characterizes visceral epithelial cells of rat glomeruli. Am J Pathol 149:319–327
Yu PT, Babicky M, Jaquish D, French R, Marayuma K, Mose E, Niessen S, Hoover H, Shields D, Cheresh D, Cravatt BF, Lowy AM (2012) The RON-receptor regulates pancreatic cancer cell migration through phosphorylation-dependent breakdown of the hemidesmosome. Int J Cancer 131:1744–1754
Zernig G, Wiche G (1985) Morphological integrity of single adult cardiac myocytes isolated by collagenase treatment: immunolocalization of tubulin, microtubule-associated proteins 1 and 2, plectin, vimentin, and vinculin. Eur J Cell Biol 38:113–122
Zhang T, Haws P, Wu Q (2004) Multiple variable first exons: a mechanism for cell- and tissue-specific gene regulation. Genome Res 14:79–89
Acknowledgments
We thank Irmgard Fischer and Rocio G. Valencia for help in preparing figures. The authors’ work was supported by the Austrian Science Research Fund, the Dystrophic Epidermolysis Bullosa Research Association (Debra, UK), and NanoNet COST Action (BM 1002).
Author information
Authors and Affiliations
Corresponding author
Additional information
Maria J. Castañón, Gernot Walko and Lilli Winter contributed equally to this review.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Castañón, M.J., Walko, G., Winter, L. et al. Plectin–intermediate filament partnership in skin, skeletal muscle, and peripheral nerve. Histochem Cell Biol 140, 33–53 (2013). https://doi.org/10.1007/s00418-013-1102-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00418-013-1102-0