
Abstract
Burosumab, a fully human monoclonal antibody to FGF23, is the only approved treatment for X-linked hypophosphatemia (XLH), a rare genetic disorder characterized by renal phosphate wasting and substantial cumulative musculoskeletal morbidity. During an initial 24-week randomized, controlled trial, 134 adults with XLH received burosumab 1 mg/kg (n = 68) or placebo (n = 66) every 4 weeks. After 24 weeks, all subjects received open-label burosumab until week 48. This report describes the efficacy and safety of burosumab during the open-label treatment period. From weeks 24–48, serum phosphorus concentrations remained normal in 83.8% of participants who received burosumab throughout and were normalized in 89.4% who received burosumab after placebo. By week 48, 63.1% of baseline fractures/pseudofractures healed fully with burosumab, compared with 35.2% with burosumab after placebo. In both groups, burosumab was associated with clinically significant and sustained improvement from baseline to week 48 in scores for patient-reported outcomes of stiffness, pain, physical function, and total distance walked in 6 min. Rates of adverse events were similar for burosumab and placebo. There were no fatal adverse events or treatment-related serious adverse events. Nephrocalcinosis scores did not change from baseline by more than one grade at either week 24 or 48. These data demonstrate that in participants with XLH, continued treatment with burosumab is well tolerated and leads to sustained correction of serum phosphorus levels, continued healing of fractures and pseudofractures, and sustained improvement in key musculoskeletal impairments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
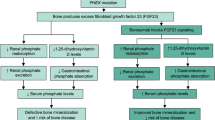
X-linked hypophosphatemia (XLH) is a rare genetic disorder with substantial musculoskeletal and functional morbidities. XLH results from loss-of-function mutations in the PHEX gene (phosphate-regulating endopeptidase homologue, X-linked), causing increased circulating levels of fibroblast growth factor 23 (FGF23) [1, 2]. Excess FGF23 impairs renal phosphate reabsorption and reduces serum 1,25-dihydroxyvitamin D (1,25(OH)2D) [3], resulting in chronic hypophosphatemia, rickets, and osteomalacia. These abnormalities lead to bone deformities, fractures and pseudofractures, and bone and joint pain. Over time, progressive enthesopathy results in pain, stiffness, and decreased mobility [4, 5]. A majority of adults with XLH also have frequent dental abscesses leading to tooth loss [5].
For decades, the standard of care for children and symptomatic adults with XLH has been to administer oral phosphate supplements and active vitamin D metabolites or analogs [6]. Management of comorbidities included treatment of pain and joint stiffness and rigorous oral hygiene. However, none of these therapies addresses the underlying pathophysiology of XLH. Furthermore, treatment with phosphate and vitamin D is often complicated by secondary hyperparathyroidism and nephrocalcinosis [7,8,9].
Burosumab (previously KRN23) is a recombinant, fully human IgG1 monoclonal antibody that binds circulating FGF23 and inhibits its excess activity [10, 11]. Based on results from phase 1, 2, and 3 trials [10,11,12,13], in 2018 burosumab became the first drug approved for the treatment of XLH in adults and children ≥ 1 year of age in the US and Canada and in children ≥ 1 year of age in the European Union. In the double-blind placebo-controlled period of a phase 3 trial in adults with XLH, administration of burosumab every 4 weeks for 24 weeks was associated with rapid normalization of serum phosphorus concentration, an approximately 17-fold greater likelihood of fracture healing, and a significant reduction in stiffness compared with placebo [13]. Reductions in pain and physical function impairment were also observed from baseline to week 24 in the burosumab group.
In that trial, after completion of the 24-week, placebo-controlled period, participants transitioned to a 24-week open-label burosumab treatment continuation period, in which efficacy and safety assessments were continued, followed by an ongoing open-label extension period for up to two additional years. The present report summarizes the efficacy of burosumab administration in each group during the treatment continuation period and analyzes all available safety data throughout the duration of burosumab exposure, which ranged from 24 to 74 weeks.
Methods
Study Design
Complete methods have been published for the initial 24-week randomized, double-blind, placebo-controlled period of this phase 3 trial [13], which was conducted at 25 centers in the US, UK, Japan, France, South Korea, Ireland, and Italy. Eligible participants were randomized 1:1 to receive burosumab or placebo in a double-blind fashion administered subcutaneously every 4 weeks for 24 weeks. Thereafter, participants from both groups entered a treatment continuation period during which all participants received open-label burosumab at 1 mg/kg every 4 weeks.
Participants
Key inclusion criteria were age 18 to 65 years and a diagnosis of XLH supported by a confirmed PHEX mutation in the participant or related family member. PHEX mutation analysis was performed by a central laboratory using guidelines set forth by the American College of Medical Genetics [14], in silico predictor models, and publicly available databases to determine pathogenicity. Clinical and laboratory data at screening were consistent with a diagnosis of XLH: serum phosphorus concentration below the lower limit of normal (LLN, 2.5 mg/dL [0.81 mmol/L]) and tubular maximum for phosphate reabsorption per glomerular filtration rate (TmP/GFR) below 2.5 mg/dL (0.81 mmol/L), as measured at a local laboratory. A Brief Pain Inventory (BPI) Worst Pain score of ≥ 4 on a scale from 0 to 10 at screening was also required. Key exclusion criteria were corrected serum calcium ≥ 10.8 mg/dL (2.7 mmol/L), serum intact parathyroid hormone (iPTH) ≥ 2.5-fold the upper limit of normal, use of a medication to suppress parathyroid hormone within 60 days before screening, or a recent history (≤ 6 months) of traumatic fracture or orthopedic surgery. Potential participants taking oral phosphate and active vitamin D metabolites or analogs were screened, and if found eligible, enrolled after a 2-week washout period.
The study was designed, conducted, recorded, and reported in accordance with the principles established by the World Medical Association Declaration of Helsinki Ethical Principles for Medical Research Involving Human Subjects. The Institutional Review Board or Ethics Committee for each site approved the study protocol. Investigators obtained written informed consent from each study participant. The clinical trial was registered as NCT02526160/EudraCT2014-005529-11.
Treatments
From baseline to week 24, participants received either burosumab 1 mg/kg (rounded to the nearest 10 mg, maximum 90 mg) or matching placebo, administered subcutaneously every 4 weeks. After week 24, all participants received burosumab. Through week 48, trained healthcare providers administered all study injections. Although treatment in weeks 24–48 was not blinded, participants and study site staff remained blinded to the original treatment assignment. In this report, the treatment groups are described as “burosumab–burosumab” or “placebo–burosumab” to describe the sequence of treatments received before and after week 24. If the serum phosphorus concentration was greater than 5.0 mg/dL (1.61 mmol/L) at any time or if two sequential serum phosphorus concentrations were between 4.5 and 5.0 mg/dL, the dose of burosumab was decreased by half; subsequent dose adjustments were determined by the investigator and medical monitor.
Outcomes
Efficacy assessments were fasting measurements of serum phosphorus (weeks 0, 1, 2, 4, 6, 10, 12, 14, 18, 20, 21, 22, 24, 26, 28, 34, 36, 46, and 48), serum 1,25(OH)2D (weeks 0, 1, 2, 4, 20, 21, 22, 24, 46, and 48), and TmP/GFR (weeks 0, 2, 4, 12, 22, 24, and 48). Estimated GFR was calculated by using the Chronic Kidney Disease Epidemiology Collaboration equation [15]. Biochemical markers of bone remodeling were serum procollagen type 1 N-propeptide (PINP), carboxy-terminal cross-linked telopeptide of type I collagen (CTX), and bone-specific alkaline phosphatase (every 12 weeks). Patient-reported outcomes were assessed using the short-form BPI questionnaire [16] and the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) questionnaire [17] (every 12 weeks). Efficacy endpoints for patient-reported outcomes were BPI Worst Pain, scored from 0 (no pain) to 10 (worst pain), and the normalized WOMAC scores for stiffness and physical function, scored from 0 (best health state) to 100 (worst health state). Functional exercise capacity was measured as total distance walked during the 6-min walk test [18] (every 12 weeks). Active, unhealed fractures (visible fracture line extending across the entire cortex) and pseudofractures (visible fracture line involving a portion of the cortex) were identified at baseline by a complete radiographic skeletal survey; radiographs at baseline fracture sites were repeated at 12-week intervals to assess healing. Partial healing was defined as a fracture line that was partially visible and partially obscured by callous formation; full healing was defined as focal cortical thickening/contour deformity with no evidence of an active fracture line. Safety measurements included adverse events and serious adverse events (at any time), renal ultrasound nephrocalcinosis grades, fasting serum calcium (same schedule as phosphorus) and plasma iPTH (same schedule as 1,25(OH)2D), and 24-h urine calcium excretion (every 12 weeks). All postbaseline serum and plasma concentrations were determined by a central laboratory. Ultrasonographic findings of nephrocalcinosis were graded on a 5-point scale, as follows: 0 = normal, 1 = faint hyperechogenic rim around the medullary pyramids, 2 = more intense echogenic rim with echoes faintly filling the entire pyramid, 3 = uniformly intense echoes throughout the pyramid, and 4 = stone formation: solitary focus of echoes at the tip of the pyramid [8, 19].
Statistical analysis
The primary and key secondary study endpoints compared the efficacy of burosumab and placebo through week 24; the analytical methods were described [13] (also see Supplemental Methods). Secondary and exploratory endpoints for the treatment continuation period included the changes from baseline through week 48 for each efficacy measurement, and the number and percentage of pseudofractures/fractures identified at baseline that were fully or partially healed at weeks 12, 24, 36, and 48. Safety endpoints included incidences and exposure-adjusted incidences for adverse events throughout the duration of burosumab exposure, which ranged from 24 to 74 weeks.
Descriptive summaries for efficacy and safety results were provided by treatment group. For selected endpoints, the least squares (LS) mean, standard error (SE), and 95% confidence interval (CI) for the change from baseline to week 48 were determined for each treatment group using generalized estimating equation (GEE) repeated-measures analysis, including treatment group, actual stratification factor, visit, and interaction of group-by-visit as fixed factors, adjusted for baseline measurement. Similar GEE models were used in the placebo–burosumab group to examine the LS mean change from weeks 24 to 48 for BPI Worst Pain, WOMAC physical function and stiffness, and total distance walked in 6 min. Probabilities of full healing of fractures/pseudofractures in each group at week 48 were estimated using a generalized linear mixed model for binomial distribution that included treatment group, visit, interaction of group-by-visit, and fracture type as fixed factors, accounting for nesting of fractures within participants. Two-sided hypothesis tests were performed at the 5% level of significance.
Results
Participants and Treatment
One hundred thirty-four participants were enrolled and randomized 1:1 to receive burosumab (n = 68) or placebo (n = 66). One-hundred thirty-three participants completed the 24-week placebo-controlled period and entered the treatment continuation period, and one participant in the burosumab–burosumab group withdrew consent after approximately 6 months (Fig. 1). Overall, 126 (94.0%) participants completed week 48 of the treatment continuation period and entered the open-label extension. Seven participants discontinued between weeks 24 and 48; one withdrew consent, one became pregnant, and five either chose to discontinue or did not re-consent after the study protocol was amended in July and September 2016. No participant discontinued the study due to an adverse event. All participants (n = 134) were included in the efficacy and safety analyses.

Disposition of study participants
Baseline demographic, clinical, and radiographic characteristics were similar between the treatment groups (Table 1). The mean age of participants was 40 years and 87 (64.9%) were female. Seventy (52.2%) participants had a total of 156 fractures (n = 27) or pseudofractures (n = 129), predominately in the weight-bearing bones of the lower extremities. Osteoarthritis was present in 63.4% of participants, and enthesopathy was present in nearly all participants (99.3%). We found no association between the presence of fractures/pseudofractures and participant age, sex, body mass index, serum phosphorus concentration, or bone turnover markers at baseline. Patient-reported outcome scores (mean ± SD) at baseline were as follows: WOMAC stiffness, 63.1 ± 20.50; WOMAC physical function, 47.4 ± 20.02; and BPI Worst Pain, 6.7 ± 1.37. At screening, all participants reported BPI Worst Pain score of 4 (moderate pain) or greater, per the study eligibility criteria, and most (71.6%) reported BPI Worst Pain score of > 6.0 (severe pain) at baseline. For the entire group at baseline, severe or extreme stiffness (a score of 3 or 4, respectively; scale 0–4) upon awakening, and later in the day, was reported by 79 (59.0%) and 68 (50.7%) participants, respectively. Stiffness scores did not associate with age, sex, presence of enthesopathy or osteoarthritis, or serum phosphorus concentration at baseline.
Before enrolling in the current study, most participants (90.3%) had received oral phosphate or vitamin D metabolites/analogs and the majority (69.4%) had first received these therapies in childhood (Online Resource 1). The mean duration of therapy was 16.5 years for phosphate and 18.2 years for active vitamin D. At baseline, 67.9% of participants were using pain medication; 22.4% used an opioid.
Pharmacodynamic Effects of Burosumab
Burosumab-Burosumab Group
In participants who received burosumab throughout the study, mean serum phosphorus concentrations increased to values above the LLN at week 1 and remained at or above this value throughout, when measured both at the midpoint (Fig. 2a) and the end (Fig. 2b) of each 4-week dosing interval. Mean phosphorus concentrations (averaged across the midpoints) were above the LLN in 94.1% of participants between weeks 0 and 24, and in 83.8% between weeks 24 and 48 (Online Resource 2). The LS mean (95% CI) increase in phosphorus concentrations from baseline to week 46 (midpoint of dosing interval) was 0.9 mg/dL (0.7, 1.0) and from baseline to week 48 (end of dosing interval) was 0.4 mg/dL (0.2, 0.5). The increase was attributed to the increase in renal phosphate reabsorption, as reflected by the LS mean (95% CI) increase from baseline to week 48 in TmP/GFR (0.5 mg/dL [0.3, 0.7]; Fig. 2c). LS mean (95% CI) increases from baseline to week 48 were also seen for serum 1,25(OH)2D (7.1 pg/mL [1.5, 12.8]; Fig. 2d), PINP (35.7 ng/mL [17.5, 53.9]; Fig. 2e), CTX (127.1 pg/mL [36.7, 217.5]; Fig. 2f), and BALP (1.2 µg/L [− 2.3, 4.8]; Fig. 2g). For each laboratory value, the largest increase was seen at the first post-baseline measurement, with subsequent attenuation and stabilization of the effect of burosumab over the course of the study.

Effect of burosumab on concentrations of a serum phosphorus measured at the midpoint of the dosing interval (1–2 weeks after a dose), b serum phosphorus measured at the end of the dosing interval (4 weeks after a dose), c TmP/GFR (tubular maximum for phosphate reabsorption per glomerular filtration rate) (TmP/GFR), d serum 1,25-dihydroxyvitamin D [1,25(OH)2D], e procollagen type 1 N-propeptide (PINP), f carboxy-terminal cross-linked telopeptide of type I collagen (CTX), and g bone-specific alkaline phosphatase (BALP). Participants received either burosumab or placebo in a double-blind fashion every 4 weeks for the first 24 weeks; all participants then received burosumab every 4 weeks beginning at week 24 while remaining blinded to their previous treatment assignment. Arrows indicate administration of study drug (burosumab or placebo). Blood and urine were collected after a minimum overnight fasting time of 8 h and prior to study drug administration. Data are expressed as mean ± standard error. The dashed lines are the lower and upper limits of normal. Conversion factors: phosphorus and TmP/GFR, 1 mg/dL = 0.32 mmol/L; 1,25(OH)2D, 1 pg/mL = 2.6 pmol/L
Placebo–Burosumab Group
During administration of placebo from weeks 0 to 24, little or no effect on serum phosphorus, TmP/GFR, or bone remodeling markers was observed (Fig. 2). After participants initiated burosumab at week 24, serum phosphorus concentrations promptly increased and remained at or above the LLN, with LS mean (95% CI) increases from baseline seen through week 46 (midpoint of dosing interval; 1.0 mg/dL [0.8, 1.1]; Fig. 2a) and week 48 (end of dosing interval; 0.4 mg/dL [0.2, 0.6]; Fig. 2b). Mean phosphorus concentrations (measured at midpoints) were above the LLN in 7.6% of participants during administration of placebo (weeks 0–24), and in 89.4% of participants during administration of burosumab (weeks 24–48) (Online Resource 2). LS mean (95% CI) increases from baseline to week 48 were also seen for 1,25(OH)2D (10.2 pg/mL [4.7, 15.7]; Fig. 2c), TmP/GFR (0.6 mg/dL [0.4, 0.8]; Fig. 2d), PINP (76.9 ng/mL [54.9, 98.9]; Fig. 2e), CTX (295.9 pg/mL [200.8, 391.1]; Fig. 2f), and BALP (6.7 µg/L [2.6, 10.7]; Fig. 2g) after initiation of burosumab. As in the other treatment group, the largest increase for each laboratory value was seen at the first assessment after initiation of burosumab at week 24.
Fracture/Pseudofracture Healing
Burosumab–Burosumab Group
At baseline in the burosumab–burosumab group, 32 (47.1%) participants had a total of 65 unhealed fractures (n = 14) or pseudofractures (n = 51). The proportion of fractures fully healed in this group at weeks 12, 24, 36, and 48 was 20.0, 43.1, 50.8, and 63.1%, respectively (Fig. 3). Probabilities of full healing estimated from a generalized linear mixed model are provided in Online Resource 2. At week 48, 80.0% of baseline fractures and pseudofractures were either fully (63.1%) or partially (16.9%) healed; 9.2% were unhealed and 10.8% were not evaluable because follow-up X-rays were not obtained. Full and partial healing by anatomic location are summarized in Online Resource 3.

Proportion of baseline fractures and pseudofractures fully healed. Participants received either burosumab or placebo in a double-blind fashion every 4 weeks for the first 24 weeks; all participants then received burosumab every 4 weeks beginning at week 24 while remaining blinded to their previous treatment assignment
Placebo–Burosumab Group
At baseline in the placebo–burosumab group, 38 (57.6%) participants had a total of 91 unhealed fractures (n = 13) or pseudofractures (n = 78). During placebo administration, only 7.7% of fractures and pseudofractures were fully healed at weeks 12 and 24 (Fig. 3). After transition to burosumab, 23.1% and 35.2% of fractures and pseudofractures were fully healed at weeks 36 and 48, respectively, mirroring the rates of healing at weeks 12 and 24 in the burosumab–burosumab group. Probabilities of full healing estimated from a generalized linear mixed model are provided in Online Resource 2. At week 48, 74.8% of baseline fractures and pseudofractures were either fully (35.2%) or partially (39.6%) healed; 12.1% were unhealed and 13.2% were not evaluable because follow-up X-rays were not obtained. Full and partial healing by anatomic location are summarized in Online Resource 3.
Patient-Reported Outcomes and Functional Exercise Capacity
Burosumab–Burosumab Group
Burosumab for 48 weeks was associated with a sustained improvement in scores for the patient-reported outcomes of stiffness, pain, and physical function, and improvement in total distance walked in 6 min (Fig. 4, Online Resource 2). The improvements from baseline to week 48 were significant (p < 0.001) for WOMAC stiffness (LS mean ± SE: − 16.03 ± 3.315) and WOMAC physical function impairment (− 7.76 ± 2.146), where lower scores represent better health. Severe or extreme stiffness upon waking (WOMAC Question 6) was reported by 63% of participants at baseline and decreased to 45% at week 24 and only 27% at week 48. Severe or extreme stiffness later in the day (WOMAC Question 7) was reported by 54% of participants at baseline and decreased to 31% at week 24 and 26% at week 48 (Online Resource 4). Improvements from baseline to week 48 were also observed for BPI Worst Pain score (LS mean ± SE: − 1.09 ± 0.216) and total distance walked in 6 min (30.5 ± 6.93 m) (both p < 0.001) (Fig. 4). Opioids were used by 25.0% of participants at baseline, 23.5% at week 24, and 20.6% at week 48; this change was not statistically significant.

Effect of burosumab on patient-reported outcomes and function, including a Western Ontario McMaster Universities Osteoarthritis Index (WOMAC) stiffness, b WOMAC physical function, c Brief Pain Inventory (BPI) Worst Pain, and d total distance walked in 6 min. Participants received either burosumab or placebo in a double-blind fashion every 4 weeks for the first 24 weeks; all participants then received burosumab every 4 weeks beginning at week 24 while remaining blinded to their previous treatment assignment. Data are expressed as mean ± standard error. *p < 0.001 for least squares mean change from baseline to week 48 (burosumab–burosumab), **p < 0.001 for least squares mean change from weeks 24 to 48 (placebo–burosumab)
Placebo–Burosumab Group
Participants receiving placebo reported modest improvements in pain but no improvement in scores for stiffness and physical function at week 24 (Fig. 4). After transition to burosumab, participants reported significant reductions from weeks 24 to 48 for WOMAC stiffness (LS mean ± SE: − 15.82 ± 2.795) and WOMAC physical function impairment (LS mean ± SE: − 8.18 ± 1.716; both p < 0.0001), where lower scores represent better health (Online Resource 2). Specifically, severe or extreme stiffness upon waking (WOMAC Question 6) was reported by 54% of participants at baseline and week 24 but fell to 29% at week 48. Severe or extreme stiffness later in the day (WOMAC Question 7) was reported by 47% of participants at baseline, 42% at week 24, but by only 20% at week 48 (Online Resource 4). Participants also reported that BPI Worst Pain decreased significantly from week 24 to week 48 (LS mean ± SE: − 1.18 ± 0.216; p < 0.001; Fig. 4). In the placebo–burosumab group, opioids were used by 19.7% of participants at baseline, 21.2% at week 24, and 17.5% at week 48; these changes were not statistically significant. Additionally, scores for functional exercise capacity (total distance walked in 6 min), which did not change from baseline to week 24 in this group, increased from weeks 24 to 48 (LS mean ± SE: 23.0 ± 5.62 m, p < 0.001; Fig. 4). Of note, participants and study staff remained blinded to the initial treatment assignment through week 48.
Safety
Safety data were obtained throughout the period of burosumab exposure, including the extension period. The mean duration of exposure in the burosumab–burosumab group was 404 days (range 167–521) and in the placebo–burosumab group, 242 days (range 165–403). The incidence, nature, and severity of adverse events were similar between treatment groups (Table 2). There were no treatment-related serious adverse events, study or treatment discontinuations due to an adverse event, or fatal adverse events. Exposure-adjusted rates of individual adverse events that were reported during exposure to burosumab were similar to those reported during exposure to placebo (Online Table 5).
Nephrocalcinosis grades of 1–3 were observed at baseline in 73 (54.5%) participants. At week 24, nephrocalcinosis increased by one grade in 11 (16.2%) participants receiving burosumab and 12 (18.2%) participants receiving placebo; by week 48, nephrocalcinosis grades decreased to baseline levels in the majority of these participants (6 of 11 [54.5%] in the burosumab–burosumab group and 8 of 12 [66.7%] in the placebo–burosumab group). Nephrocalcinosis decreased by one grade in 8 (11.8%) participants receiving burosumab (4 at week 24 and 4 at week 48) and in 5 (7.6%) participants in the placebo–burosumab group (4 at week 24 and 1 at week 48). Nephrocalcinosis did not change from baseline by more than one grade in any participant, and changes in nephrocalcinosis grade were not associated with changes in urine calcium excretion, serum iPTH, or estimated GFR.
Due to increases in serum phosphorus concentration above the target range, the dose of burosumab was decreased in 5 (7.4%) participants in the burosumab–burosumab group (during weeks 0–24) and 4 (6.1%) participants in the placebo–burosumab group (during weeks 24–48); phosphorus levels subsequently normalized in each participant. One of these participants subsequently received burosumab at the original dose of 1 mg/kg. Mean serum calcium, plasma iPTH, and urine calcium excretion did not increase with burosumab administration (Fig. 5).

Effect of burosumab on a serum calcium, b 24-h urine calcium excretion, and c plasma intact parathyroid hormone (iPTH). Participants received either burosumab or placebo in a double-blind fashion every 4 weeks for the first 24 weeks; all participants then received burosumab every 4 weeks beginning at week 24 while remaining blinded to their previous treatment assignment. Arrows indicate administration of study drug (burosumab or placebo). Blood and urine were collected after a minimum overnight fasting time of 8 h and prior to study drug administration. Data are expressed as mean ± standard error. The dashed lines are the lower and upper limits of normal
Two female participants discontinued the study due to pregnancy; one during the treatment continuation period, as described above, and one after completing the treatment continuation period during the open-label extension. Each woman carried the pregnancy to term and delivered a healthy infant with no complications.
Discussion
In the present 48-week, phase 3 trial of burosumab, adults with XLH participated in a 24-week randomized, double-blind, placebo-controlled period to evaluate the efficacy and safety of burosumab and a subsequent 24-week continuation period in which all participants received burosumab to confirm efficacy and safety with longer-term use. The results demonstrate that with continued exposure to burosumab, phosphorus homeostasis was maintained, the proportion of fully healed fractures and pseudofractures progressively increased, and pain, stiffness, physical function, and functional exercise capacity scores improved. Despite long-standing physical impairments from a lifetime of XLH, participants experienced clinically meaningful benefits that began shortly after burosumab was initiated and persisted with continued administration, with an overall positive benefit-risk profile through at least 48 weeks of treatment.
In the initial 24 weeks, administration of burosumab resulted in significant improvements in serum phosphorus, TmP/GFR, 1,25(OH)2D, bone biomarkers, fracture healing, and patient-reported outcomes, compared with administration of placebo [13]. In the subsequent 24 weeks when all participants received burosumab, mean serum phosphorus concentration, measured at the midpoints of dose intervals, was within the normal range in 84% and 89% of participants in the burosumab–burosumab and placebo–burosumab groups, respectively, demonstrating sustained efficacy of burosumab in normalizing phosphorus homeostasis. In each group, the maximum effects on serum phosphorus and 1,25(OH)2D were observed after the first dose of burosumab, with a small attenuation in the drug’s effect with subsequent doses. After three or four doses, serum phosphorus remained stable and within the normal range, which is the therapeutic target for burosumab. The relative long-term stability of serum phosphorus and 1,25(OH)2D suggests that desensitization to the effects of burosumab does not occur over time.
In patients with XLH, chronic osteomalacia can lead to fractures and pseudofractures, which were observed in half of the study participants at baseline, most commonly in the lower extremities. The proportion of fractures and pseudofractures that healed was greater over time in the group receiving burosumab, with 80% achieving partial or complete healing by week 48. For the group initiating burosumab at week 24, healing of fractures and pseudofractures at week 48 replicated the healing at week 24 in the group receiving burosumab from the outset. Healing of fractures and pseudofractures with burosumab is presumed to reflect an improvement in bone mineralization and remodeling. This formulation is supported by the initial increase in markers of bone formation (PINP, BALP) and resorption (CTX) after initiating burosumab in each treatment group, followed by a gradual attenuation of the increase in bone biomarkers over time.
All participants reported at least moderate pain, which was a criterion for enrollment, with a large majority reporting severe pain. A majority of participants also reported severe or extreme stiffness. Mean scores for stiffness and physical function were similar to or worse than in patients with severe osteoarthritis [20]. After initiation of burosumab, scores for pain, stiffness, and physical function improved significantly from baseline in both treatment groups. After 24 weeks of burosumab, the magnitude of improvement was almost identical in participants initiating burosumab at week 0 and in those transitioning to burosumab at week 24. By week 48, fewer than 30% of participants in either group had residual severe or extreme stiffness. Functional exercise capacity, as measured by the total distance walked in 6 min, also improved after initiation of burosumab in each group. The consistent effects on patient-reported outcomes during the first 24 weeks of burosumab and the sustained effects through week 48 are notable and provide further evidence of the benefits of burosumab in XLH patients previously treated with conventional therapy. These improvements were achieved despite long-standing skeletal abnormalities that can cause pain that is not lessened by healing of osteomalacia or fractures. A majority of participants had osteoarthritis at baseline, and almost all had enthesopathy on baseline radiographs. It remains to be seen whether normalization of serum phosphorus with burosumab affects the incidence or course of enthesopathy. Together, these findings suggest that treatment with burosumab may well be associated with continued improvement in the substantial burden of disease that accumulates over a lifetime, improvements that are clinically meaningful to adults with XLH.
The present analysis identified no new safety concerns relative to those previously observed [13]. Adverse events observed during administration of burosumab were similar to those during administration of placebo, and the adverse event profile did not appear to change with an additional 24 weeks of burosumab. Most adverse events were mild to moderate in severity, and no participant stopped burosumab or withdrew from the study due to an adverse event. No treatment-related serious adverse events, deaths, or life-threatening adverse events were observed. Elevation in serum phosphorus concentration, observed in 7% of participants during burosumab treatment, was without clinical manifestations and resolved after dose reduction. Nephrocalcinosis and hyperparathyroidism are known complications associated with oral phosphate and active vitamin D [6, 7, 9, 21]. No participant had more than a single grade increase or decrease in nephrocalcinosis score. Serum calcium and PTH concentrations and urine calcium excretion were unchanged with burosumab.
These data demonstrate that in adults with XLH, treatment with burosumab is associated with significant efficacy and an acceptable safety profile through 48 weeks. Sustained normalization of serum phosphorus concentration leads to healing of fractures and pseudofractures, strongly suggesting that bone mineralization and remodeling also improve with continued burosumab treatment. Use of the novel agent burosumab to normalize phosphorus homeostasis in adults with XLH results in sustained improvement in the important clinical parameters of stiffness, pain, and functional exercise capacity.
References
The HYP Consortium (1995) A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. Nat Genet 11:130–136. https://doi.org/10.1038/ng1095-130
Jonsson KB, Zahradnik R, Larsson T, White KE, Sugimoto T, Imanishi Y, Yamamoto T, Hampson G, Koshiyama H, Ljunggren O, Oba K, Yang IM, Miyauchi A, Econs MJ, Lavigne J, Juppner H (2003) Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med 348:1656–1663. https://doi.org/10.1056/nejmoa020881
Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T (2004) FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res 19:429–435. https://doi.org/10.1359/jbmr.0301264
Hardy DC, Murphy WA, Siegel BA, Reid IR, Whyte MP (1989) X-linked hypophosphatemia in adults: prevalence of skeletal radiographic and scintigraphic features. Radiology 171:403–414. https://doi.org/10.1148/radiology.171.2.2539609
Chesher D, Oddy M, Darbar U, Sayal P, Casey A, Ryan A, Sechi A, Simister C, Waters A, Wedatilake Y, Lachmann RH, Murphy E (2018) Outcome of adult patients with X-linked hypophosphatemia caused by PHEX gene mutations. J Inherit Metab Dis 41:865–876. https://doi.org/10.1007/s10545-018-0147-6
Linglart A, Biosse-Duplan M, Briot K, Chaussain C, Esterle L, Guillaume-Czitrom S, Kamenicky P, Nevoux J, Prie D, Rothenbuhler A, Wicart P, Harvengt P (2014) Therapeutic management of hypophosphatemic rickets from infancy to adulthood. Endocr Connect 3:R13–30. https://doi.org/10.1530/ec-13-0103
Rivkees SA, el-Hajj-Fuleihan G, Brown EM, Crawford JD (1992) Tertiary hyperparathyroidism during high phosphate therapy of familial hypophosphatemic rickets. J Clin Endocrinol Metab 75:1514–1518. https://doi.org/10.1210/jcem.75.6.1464657
Verge CF, Lam A, Simpson JM, Cowell CT, Howard NJ, Silink M (1991) Effects of therapy in X-linked hypophosphatemic rickets. N Engl J Med 325:1843–1848. https://doi.org/10.1056/nejm199112263252604
Taylor A, Sherman NH, Norman ME (1995) Nephrocalcinosis in X-linked hypophosphatemia: effect of treatment versus disease. Pediatr Nephrol 9:173–175
Carpenter TO, Imel EA, Ruppe MD, Weber TJ, Klausner MA, Wooddell MM, Kawakami T, Ito T, Zhang X, Humphrey J, Insogna KL, Peacock M (2014) Randomized trial of the anti-FGF23 antibody KRN23 in X-linked hypophosphatemia. J Clin Investig 124:1587–1597. https://doi.org/10.1172/jci72829
Imel EA, Zhang X, Ruppe MD, Weber TJ, Klausner MA, Ito T, Vergeire M, Humphrey JS, Glorieux FH, Portale AA, Insogna K, Peacock M, Carpenter TO (2015) Prolonged correction of serum phosphorus in adults with X-linked hypophosphatemia using monthly doses of KRN23. J Clin Endocrinol Metab 100:2565–2573. https://doi.org/10.1210/jc.2015-1551
Ruppe M, Peacock M, Weber T, Portale A, Insogna K, Imel E, Luca D, Skrinar A, Mealiffe M, San Martin J, Carpenter T (2016) Clinical and radiographic characteristics of adult X-linked hypophosphatemia (XLH) in a cohort of patients treated with KRN23, an antibody to FGF23. In: Presented at: ASBMR 2016 annual meeting, 16–19 September 2016, Atlanta, Georgia Presentation, #MO0319
Insogna KL, Briot K, Imel EA, Kamenicky P, Ruppe MD, Portale AA, Weber T, Pitukcheewanont P, Cheong HI, Jan de Beur S, Imanishi Y, Ito N, Lachmann RH, Tanaka H, Perwad F, Zhang L, Chen CY, Theodore-Oklota C, Mealiffe M, San Martin J, Carpenter TO (2018) A randomized, double-blind, placebo-controlled, phase 3 trial evaluating the efficacy of burosumab, an anti-FGF23 antibody, in adults with X-linked hypophosphatemia: Week 24 primary analysis. J Bone Miner Res 33:1383–1393. https://doi.org/10.1002/jbmr.3475
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424. https://doi.org/10.1038/gim.2015.30
Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF III, Feldman HI, Kusek JW, Eggers P, Van Lente F, Greene T, Coresh J, CKD EPI (2009) A new equation to estimate glomerular filtration rate. Ann Intern Med 150:604–612
Daut RL, Cleeland CS, Flanery RC (1983) Development of the Wisconsin Brief Pain Questionnaire to assess pain in cancer and other diseases. Pain 17:197–210
Bellamy N (2012) WOMAC osteoarthritis index user guide. Version X. Brisbane
ATS statement: guidelines for the six-minute walk test (2002). Am J Respir Crit Care Med 166:111–117. https://doi.org/10.1164/ajrccm.166.1.at1102
Patriquin H, Robitaille P (1986) Renal calcium deposition in children: sonographic demonstration of the Anderson–Carr progression. Am J Roentgenol 146:1253–1256. https://doi.org/10.2214/ajr.146.6.1253
Angst F, Aeschlimann A, Michel BA, Stucki G (2002) Minimal clinically important rehabilitation effects in patients with osteoarthritis of the lower extremities. J Rheumatol 29:131–138
Carpenter TO, Imel EA, Holm IA, Jan de Beur SM, Insogna KL (2011) A clinician’s guide to X-linked hypophosphatemia. J Bone Miner Res 26:1381–1388. https://doi.org/10.1002/jbmr.340
Acknowledgements
The authors acknowledge the substantial contributions of Mary D. Ruppe to this work before her death. The authors thank the participants, caregivers, and health care professionals who participated in this study. The authors also acknowledge Jonathan Latham of PharmaScribe, LLC (on behalf of Ultragenyx Pharmaceutical, Inc.) for medical writing assistance and Chao-Yin Chen of Ultragenyx Pharmaceutical, Inc. for statistical support. This work was supported by Ultragenyx Pharmaceutical, Inc. in partnership with Kyowa Hakko Kirin Co., Ltd.
Funding
Ultragenyx Pharmaceutical, Inc. supported this work in partnership with Kyowa Hakko Kirin Co., Ltd.
Author information
Authors and Affiliations
Contributions
AAP contributed to the study design and prepared the first draft of the paper. He is guarantor. LZ was responsible for statistical analysis of the data. All authors contributed to the experimental work, revised the paper critically for intellectual content, and approved the final version. All authors agree to be accountable for the work and to ensure that any questions relating to the accuracy and integrity of the paper are investigated and properly resolved.
Corresponding author
Ethics declarations
Conflict of interest
The institution that employs AAP and FP received research grants from Ultragenyx for this and other studies. AAP reports advisory board membership and lectures for Ultragenyx. TOC reports research grants from Ultragenyx and consultancy for Clementia, Inozyme, Pharmacosmos, Kirin, and Lakeside. MLB reports Honoraria from Amgen, Bruno Farmaceutici, and Kyowa Kirin, Grants and/or Speaker Fees from Abiogen, Alexion, Amgen, Bruno Farmaceutici, Eli Lilly, Kyowa Kirin, MSD, NPS, Servier, Shire, and SPA, and consultancy for Alexion, Bruno Farmaceutici, Kyowa Kirin, Servier, and Shire. KB reports Personal Fees from Kyowa Kirin, UCB, Amgen, and Lilly. RC reports Grants and Non-financial Support from Ultragenyx. SJdB reports Research Grants, Advisory Board Membership, and Consultancy for Ultragenyx. RE reports Grants and Personal Fees from Amgen and Alexion, Grants from Department of Health, AstraZeneca, ARUK/MRC Centre for Excellence in Musculoskeletal Ageing Research, National Institute for Health Research, MRC/AZ Mechanisms of Diseases Call, and MRC, Grants, Personal Fees, and Non-financial Support from Immunodiagnostic Systems, Grants and other from National Osteoporosis Society, Grants, Personal Fees, and other from Roche, Personal Fees and other from Eli Lilly, other from European Calcified Tissue Society, IOF CSA, IBMS, and ASBMR, and Personal Fees from D-STAR and GSK Nutrition. YI reports Grants from Kyowa Hakko Kirin Co., Ltd. EAI reports Grants and Advisory Board Membership for Ultragenyx Pharmaceutical, Inc. SI reports Research Grants from Ultragenyx. NI and TK report research Grants from Kyowa Hakko Kirin. MJ reports Research Grants and Advisory Board Membership for Kyowa Kirin. PK reports research grants from Ultragenyx. RK reports Research Grants and Speaking Fees from Kyowa Kirin. FP reports Advisory Board Membership and Lectures for Ultragenyx. PP reports Research Grants from Amgen, Ultragenyx, and Shire, and employment by Ascendis Pharma, Inc. SHR reports support from Ultragenyx, Kyowa Kirin, Lilly, and Amgen/UCB for enrolling patients into clinical trials, and non-financial support from Eli Lilly. YT reports Personal Fees from Kyowa Hakko Kirin Co. Ltd., and Grants and Personal Fees from Chugai Pharmaceutical Co. Ltd. and Teijin Pharma Ltd. TJW reports Research Support, Travel Fees, and Honoraria from Ultragenyx Pharmaceutical, Inc. LZ, CTO, MM, and JSM were employees and stockholders of Ultragenyx Pharmaceutical, Inc. during the study. JSM has a Patent Pending that is jointly owned by Ultragenyx Pharmaceutical, Inc. KI reports Research Grants and Advisory Board Membership for Ultragenyx. HIC, MCS, RL, HT, and HWY have nothing to disclose.
Human and Animal Rights
All procedures performed in human participants were in accordance with the Ethical Standards of the Institutional and/or National Research Committee and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Informed Consent
Informed consent was obtained from all study participants.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Portale, A.A., Carpenter, T.O., Brandi, M.L. et al. Continued Beneficial Effects of Burosumab in Adults with X-Linked Hypophosphatemia: Results from a 24-Week Treatment Continuation Period After a 24-Week Double-Blind Placebo-Controlled Period. Calcif Tissue Int 105, 271–284 (2019). https://doi.org/10.1007/s00223-019-00568-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-019-00568-3