
Abstract
We have reported that the cell-penetrating peptide (CPP) penetratin acts as a potential absorption enhancer in oral insulin delivery systems and that this action occurs through noncovalent intermolecular interactions. However, the region-dependent role of CPPs in intestinal insulin absorption has not been clarified. To identify the intestinal region where CPPs have the most effect in increasing insulin absorption, the region-dependent action of penetratin was investigated using in situ closed intestinal loops in rats. The order of the insulin area under the insulin concentration–time curve (AUC) increase effect by l-penetratin was ileum > jejunum > duodenum > colon. By contrast, the AUC order after coadministration of insulin with d-penetratin was colon > duodenum ≥ jejunum and ileum. We also compared the effects of the l- and d-forms of penetratin, R8, and PenetraMax on ileal insulin absorption. Along with the CPPs used in this study, l- and d-PenetraMax produced the largest insulin AUCs. An absorption study using ilea pretreated with CPPs showed that PenetraMax had no irreversible effect on the intestinal epithelial membrane. The degradation of insulin in the presence of CPPs was assessed in rat intestinal enzymatic fluid. The half-life (t 1/2) of insulin increased from 14.5 to 23.7 and 184.7 min in the presence of l- and d-PenetraMax, respectively. These enzymatic degradation-resistant effects might contribute partly to the increased ileal absorption of insulin induced by d-PenetraMax. In conclusion, this study demonstrated that the ability of the l- and d-forms of penetratin to increase intestinal insulin absorption was maximal in the ileum and the colon, respectively, and that d-PenetraMax is a powerful but transient enhancer of oral insulin absorption.
Similar content being viewed by others
INTRODUCTION
Insulin is a universal clinical drug used in the treatment of diabetes. Current methods for insulin administration are based primarily on subcutaneous injection, which may produce peripheral hyperinsulinemia leading to hypertension and atherosclerosis (1–3). Oral delivery of insulin, if available, would be the most desirable and frequently used mode because of its convenience and high patient compliance. Oral insulin therapy is thought to be close to the physiological state because it may improve the portal insulin level and may curtail the risk of peripheral hyperinsulinemia (4). However, the oral delivery of peptide/protein drugs such as insulin is beleaguered by their poor bioavailability, which is caused primarily by susceptibility to digestion by proteolytic enzymes in the gastrointestinal (GI) tract and poor permeability through the epithelial cell layer of the intestinal mucosal membrane (5). To have therapeutic activity, orally administered insulin must be absorbed efficiently from the intestinal lumen into the circulation without being metabolized extensively in the intestine (6).
Several approaches have been used to improve the oral absorption of insulin from the GI tract, including encapsulation of insulin in nanoparticles (7,8), coating of insulin-loaded nanoparticles with protease inhibitors (9), enteric coating of insulin-loaded nanoparticles (10), and hydrogels (11). Although some permeation enhancers can facilitate the poor permeation of these drugs through the epithelial membrane, advances in these reagents for clinical use are limited by biological efficiency at tolerable levels of safety (2). Although the susceptibility to protease digestion has been largely circumvented, the poor permeability through the intestinal mucosal layer remains the primary challenge that must be overcome to significantly augment the oral bioavailability of insulin (5,6). Consequently, a strategy to improve the absorption of peptide/protein drugs such as insulin across the intestinal epithelial barrier is essential to achieving sufficient oral bioavailability for clinical applications.
In our previous reports, we showed that, by using a noncovalent cargo-interaction strategy, cell-penetrating peptides (CPPs) can successfully deliver the most challenging peptide and protein cargos, including insulin, through the intestinal and nasal mucosae with highly effective bioavailability (12–15). Penetratin, which is derived from the Drosophila Antennapedia homeoprotein (16–18), was most successful in overcoming the barrier to the permeability of peptides and proteins through the epithelial mucosal membrane and increased the enteral insulin bioavailability by up to 35% (13). The assembly of cargo–CPP complexes via noncovalent interactions can be used to avoid the loss of function because our approach relies on the electrostatic interactions between the CPP and the peptide or protein drug under physiological conditions, whereas hydrophobic residues may contribute to hydrophobic interactions (19,20). We recently reported our findings of an in silico analysis in which we found that an innovative CPP, “PenetraMax,” developed by sequence modification of penetratin, had a superior insulin absorption-enhancing efficiency over the parent penetratin (21,22). A subchronic study of nasal administration of insulin with PenetraMax at two selected concentrations (0.5 and 2.0 mM), which significantly increased the systemic bioavailability of insulin reaching almost 100% relative to subcutaneous insulin administration, found no increase in the risk of detectable local or systemic inflammation and immunogenicity and without causing detectable epithelial membrane damage (23).
Our strategy of using new CPPs to increase oral insulin absorption is based on their ability to increase the permeation of insulin across the intestinal mucosal membrane (13,15,22). Insulin absorption appears to differ between intestinal regions because the protease content is much higher in the upper small intestine than in the descending small intestine (24). Furthermore, the permeability of macromolecular drugs is molecular weight-dependent passage over different regions of the small intestine (25). Therefore, the design of an optimal oral delivery system of insulin using a physical complex of CPPs depends on understanding the different effects of CPPs on the insulin absorption along the intestinal tract. In addition, the effects of the enzymatic degradation of insulin coadministered with the l- and d-forms of CPPs along the intestinal tract and the consequent effects on the ability of CPPs to increase insulin absorption must be investigated to clarify whether the protease inhibitory effects of CPPs affect the oral insulin bioavailability.
The purposes of our study were to investigate the effects of l- and d-penetratin on insulin absorption in different segments of the intestine (duodenum, jejunum, ileum, and colon) in the rat using an in situ loop technique and to compare the intestinal absorption-enhancing efficiency of insulin coadministered with the l- and d-forms of CPPs (R8, penetratin, and PenetraMax). We also examined whether the l- and d- forms of CPPs protected insulin against enzymatic degradation in rat intestinal fluid and measured insulin absorption in intestinal segments pretreated with CPPs to determine whether the effects of CPPs are reversible.
MATERIALS AND METHODS
Materials
Recombinant human insulin (26 IU/mg) and soybean trypsin inhibitor (STI) were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). The CPPs listed in Table I were synthesized by Sigma-Genosys, Life Science Division, of Sigma–Aldrich Japan Co. (Hokkaido, Japan). All other chemicals were of analytical grade and were commercially available.
Preparation of the Mixed Insulin–CPP Solutions
Specific amounts of insulin and CPPs were dissolved in phosphate-buffered saline (PBS; 137 mM NaCl, 2.6 mM KCl, 6.4 mM Na2HPO4·12H2O, and 1.4 mM KH2PO4 adjusted to pH 6.0). The PBS also contained 0.001% methylcellulose, which prevents the adsorption of the peptides to the plastic vial surface. To prepare the insulin solution, specific amounts of recombinant human insulin were dissolved in 100 μL of 0.1 M HCl in a polypropylene plastic vial. The insulin–HCl solution was diluted to 0.8 mL with PBS and normalized with 100 μL of 0.1 M NaOH. The insulin solution was prepared at 40 IU/mL in PBS (for a 50-IU/kg insulin dose).
Specific amounts of CPPs were measured in polypropylene plastic vials and dissolved in PBS (pH 6.0) containing 0.001% methylcellulose. The CPP solution was prepared at 1 mM in PBS. Equal volumes of insulin and CPP solutions were mixed gently at room temperature. The final concentrations of the insulin and CPP solutions were 20 IU/mL and 0.5 mM, respectively. Each insulin and CPP solution was clear after mixing.
Animals
This research was performed at Kobe Gakuin University and complied with the regulations of the Committee on Ethics in the Care and Use of Laboratory Animals. Male Sprague Dawley rats weighing 180–220 g were purchased from Japan SLC, Inc. (Shizuoka, Japan). The animals were housed in rooms with controlled temperature (23 ± 1°C) and relative humidity (55 ± 5%) and were allowed free access to water and food during acclimatization. Animals were fasted for 24 h before the experiments but were allowed to drink water ad libitum.
In Situ Closed-Loop Absorption Study
Following anesthetization by intraperitoneal (i.p.) injection of sodium pentobarbital (50 mg/kg; Somnopentyl®; Kyoritsu Seiyaku Corp., Tokyo, Japan), the rats were restrained in a supine position on a thermostatically controlled board at 37°C. Additional i.p. injections of sodium pentobarbital (12.5 mg/kg) were used every 1 h to maintain the anesthesia.
A small midline incision was made carefully in the abdomen, and 6–7-cm segments of the duodenum, jejunum, ileum, and colon were identified and cannulated at both ends using silicone tubing (5 mm OD, 3 mm ID; Sanyo Co., Ltd., Tokyo, Japan). The duodenal loop was made for the first portion of the intestine, which was the closest to the stomach. The next portion, 5 cm away from the ligament of Treitz, was used as the jejunal loop. The ileal loop was made at the end of the small intestine, just proximal to the ileocecal junction. The colonic loop was made of the ascending colon. In the experiments, the exposed segments were cannulated at both ends using silicone tubing. These segments were ligated securely to prevent fluid loss. To wash out the exposed segment content and to remove the luminal enzymes, PBS warmed to 37°C was circulated through the cannula at 5 mL/min for 4 min using an infusion pump (KD Scientific Inc., Holliston, MA, USA). Following the perfusion, the cannulation tubing was removed, and the segments were tightly closed; about 1 mL of PBS remained in the segments. The segments were returned carefully to their original location inside the peritoneal cavity. The rats were left on the board at 37°C for a further 30 min to allow recovery from the elevated blood glucose concentration caused by the surgery described above.
After the 30-min rest, 0.5 mL of insulin–CPP mixed solution or insulin solution (control) was administered directly into the 6-cm loop segment of the duodenum, jejunum, ileum, or colon. The dose of insulin was fixed at 50 IU/kg body weight (132 μM) for all absorption experiments. In the experiments designed to compare the absorption-enhancing ability of CPPs, the dose of each CPP was 1.25 μmol/kg body weight (0.5 mM). The loop was ligated securely to prevent loss of the administered solutions, and the abdominal wall was sutured to prevent heat loss. An absorption study using ileal loops pretreated with CPPs was also conducted. Following the 30-min rest, the 6-cm ileal loop was treated with 0.5 mL of l- or d-PenetraMax (0.5 mM). After 3 h of incubation, the CPP solution in the ileal loop was removed by perfusion with 20 mL of PBS warmed to 37°C at a flow rate of 5 mL/min using an infusion pump. The insulin solution was then administered into the ileal loop at a dose of 50 IU/kg.
During the experiment, a 0.25-mL blood aliquot was taken from the jugular vein using a 1-mL tuberculin heparinized syringe before and 5, 10, 15, 30, 60, 120, and 180 min after dosing. Plasma was separated by centrifugation at 12,000 rpm (13,400×g) for 1 min and stored at −80°C until analysis. The plasma insulin concentration was measured using a human insulin ELISA kit (Mercodia AB, Uppsala, Sweden), and the absorbance at λ max 450 nm was detected by using a Synergy HT microplate reader (BioTek Instruments Inc., Winooski, VT, USA).
Pharmacokinetic Analysis
The bioavailability of intestinal-administered insulin was calculated relative to the subcutaneous (s.c.) route. Briefly, an insulin solution was prepared by dissolving an appropriate amount of insulin in PBS for s.c. injection (1 IU/kg). The peak plasma concentration (Cmax) and time to reach Cmax (Tmax) were determined directly from the plasma insulin concentration–time curves. The total area under the insulin concentration–time curve (AUC) for 0–3 h was estimated from the sum of successive trapezoids between each data point. The relative bioavailability (BA) of insulin was calculated relative to the s.c. injection as follows:
Insulin Degradation in Intestinal Fluid in the Presence of CPPs
Intestinal fluid was collected from male Sprague Dawley rats by inserting a Sonde needle into the upper portion of the small intestine, and the intestine was then cannulated on the lower side (length = 20 cm) to remove the intestinal fluid. The contents of the small intestine were washed out with 20 mL of PBS. Because intestinal fluid contains a high lipid content, the efflux solution was treated with two volumes of methylene chloride to remove any lipids that might interfere with the analysis of insulin by HPLC (26). This removal of lipid contents was repeated five times. CPP (0.25 mM final concentration) was mixed with insulin (10 IU/mL final concentration) and incubated in the intestinal fluid at 37°C. At 5, 15, 30, 45, and 60 min, 50 μL was collected, and 50 μL of ice-cold mobile phase solution was added to terminate the reaction. STI (1.25 mg/mL final concentration) was used as a positive control. Insulin concentration was measured by HPLC (LaChrom Elite System, Hitachi High-Technologies Corporation, Tokyo, Japan) using the following conditions: mobile phase, acetonitrile, trifluoroacetic acid (0.1%), and sodium chloride (31:69:0.58, v/v/w); 20 μL injection volume; 1.0 mL/min flow rate; 220 nm wavelength; 4.6 × 150 mm column; 5 μm (GL-Pack Nucleosil 100-5C18, GL Science Inc., Tokyo, Japan).
Statistical Analysis
Significant differences in the mean values were evaluated by a one-way layout analysis of variance (ANOVA) followed by a Bonferroni multiple-comparison test. A p value <0.05 was considered significant.
RESULTS
Effect of l- and d-Penetratin on Insulin Absorption in the Intestinal Loops
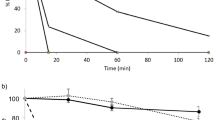
The time profiles of the plasma insulin concentration after the coadministration of insulin and l- or d-penetratin (0.5 mM) in the different intestinal segments are shown in Fig. 1. Whereas insulin alone was poorly absorbed from all intestinal regions, l- and d-penetratin increased the intestinal absorption of insulin markedly when each was coadministered with insulin into the duodenum, jejunum, ileum, and colon. The pharmacokinetic parameters derived from the insulin concentration–time profiles following in situ administration of insulin with l- or d-penetratin into different intestinal segments are shown in Table II. The AUC was derived from the plasma insulin concentration–time profiles, and relative BA was calculated from data obtained in the s.c. injection study. The largest insulin AUC was observed after coadministration of insulin with l-penetratin (0.5 mM) into ileum loops. The order of the effect of insulin AUC coadministered with l-penetratin was ileum > jejunum > duodenum > colon. By contrast, the AUC order after coadministration of insulin with d-penetratin (0.5 mM) was colon > duodenum ≥ jejunum and ileum. Treatment with 0.5 mM d-penetratin produced an insulin BA that was 13.6 times higher than that for insulin solution alone after colonic administration. By contrast, treatment with 0.5 mM l-penetratin produced an insulin BA that was 70.7 times higher than that with insulin solution alone after ileal administration (Table II).

a–d Plasma insulin concentration vs. time profiles following in situ administration of insulin (50 IU/kg) with l- or d-penetratin (0.5 mM) in various sites of the intestine. Each data point represents the mean ± SEM (n = 4–5). Multiplication signs indicate insulin–PBS solution, black circles l-penetratin, and white circles d-penetratin
Effect of CPPs on Insulin Absorption from the Ileum
Figure 2 shows the ileal absorption of insulin coadministered with either the l- or d-form of penetratin, R8, and PenetraMax. No apparent absorption was observed following administration of the insulin solution in the absence of CPP. When the l- or d-form of R8 (0.5 mM) was coadministered with insulin (50 IU/kg) into the ileal loop, insulin absorption through the ileal membrane was greater for d-R8 compared with l-R8. By contrast, l-penetratin produced a larger increase in insulin absorption compared with d-penetratin. l- and d-PenetraMax (0.5 mM) produced the greatest increase in ileal insulin absorption.

a–c Plasma insulin concentration vs. time profiles following in situ administration of insulin (50 IU/kg) with CPPs (0.5 mM) in ileal segments. Each data point represents the mean ± SEM (n = 4). Multiplication signs indicate insulin–PBS solution, black circles l-peptide, and white circles d-peptide
Table III summarizes the pharmacokinetic parameters derived from the insulin concentration–time profiles following in situ loop administration of insulin with various CPPs to the ileal segments. The coadministration of l- or d-PenetraMax significantly increased the Cmax, AUC, and BA of insulin and produced the greatest increase in ileal insulin absorption compared with the l- and d-forms of penetratin and R8. The coadministration of l- and d-PenetraMax (0.5 mM) increased the insulin AUC from 5.2 ± 0.8 to 570.2 ± 45.5 and 777.6 ± 118.1 μU h/mL, respectively. Treatment with 0.5 mM l- and d-PenetraMax resulted in a greater increase in insulin BA compared with l-penetratin by 1.5 and 2.1 times, respectively. Thus, among the CPPs used in this study, l- and d-PenetraMax showed the greatest effects on ileal insulin absorption. Therefore, l- and d-PenetraMax were used in further studies to examine the effects of pretreatment with CPPs on ileal insulin absorption.
Effect of PenetraMax Pretreatment on Insulin Absorption in the Ileum
The insulin concentration–time profiles and AUC following in situ administration of insulin into ileal segments pretreated with l- or d-PenetraMax for 3 h are shown in Figs. 3 and 4, respectively. The coadministration of l- or d-PenetraMax significantly increased ileal insulin absorption (insulin AUC 570.2 ± 45.5 and 777.6 ± 118.1 μU h/mL, respectively) as shown in Fig. 2 and Table III. By contrast, pretreatment of ileal segments with l- or d-PenetraMax had a slight effect on the insulin AUC (18.1 ± 1.9 and 29.6 ± 0.4 μU h/mL, respectively). Although the slight increase in insulin AUC following pretreatment with l- or d-PenetraMax was significant, no therapeutically effective increase in the insulin (50 IU/kg) absorption was obtained after pretreatment with and removal of l- or d-PenetraMax before insulin administration

Plasma insulin concentration vs. time profiles following in situ administration of insulin (50 IU/kg) in ileal segments coadministered (circle) or pretreated (square) for 3 h with PenetraMax (0.5 mM). Each data point represents the mean ± SEM (n = 4). Black circles and squares denote l-PenetraMax and white circles and squares d-PenetraMax

Effect of pretreatment with PenetraMax (0.5 mM) on the AUC following in situ administration of insulin (50 IU/kg) in ileal segments pretreated for 3 h. Each data point represents the mean ± SEM (n = 4). Black bars denote coadministration of PenetraMax and white bars pretreatment with PenetraMax. **p < 0.01, significantly different from the corresponding coadministration groups of PenetraMax
Insulin–CPP Stability in Intestinal Fluid
The effects of CPPs (0.25 mM) on the stability of insulin (10 IU/mL) in intestinal fluid are shown in Fig. 5. The elimination of insulin followed apparent first-order kinetics. Although insulin was rapidly degraded in intestinal enzymatic fluid, all CPPs used in this study, especially d-PenetraMax and d-penetratin, markedly reduced insulin degradation. STI was used as a positive control because it prevents the degradation of insulin by major proteases such as trypsin and chymotrypsin in rat intestinal fluid. Table IV summarizes the elimination rate constant (k el) and the half-life (t 1/2) of insulin in intestinal enzymatic fluid in the presence or absence of CPPs. The insulin k el was significantly reduced, and t 1/2 was significantly prolonged by d-penetratin (222.9 min), d-R8 (50.6 min), d-PenetraMax (184.7 min), and l-PenetraMax (23.7 min). In addition, the prolongation of insulin t 1/2 by d-penetratin, d-R8, and d-PenetraMax was greater than that by the l-form of the corresponding CPPs.

a–c Degradation profile of insulin over time in the presence of CPP (0.25 mM) in rat intestinal fluid. Each data point represents the mean ± SEM (n = 3–4). White squares indicate soybean trypsin inhibitor (STI, 1.25 mg/mL), multiplication sign insulin–PBS solution (control, 10 IU/mL), black circles l-peptide, and white circles d-peptide
DISCUSSION
Oral delivery systems for insulin as a peptide/protein model remain an attractive alternative to parenteral delivery, and the development of such systems has been a challenge. Clinicians seek methods for the oral insulin delivery for the treatment of diabetes because of its convenience and high patient compliance. However, the actual application in clinical practice has been hindered by two major obstacles: a high susceptibility of insulin to digestion by proteolytic enzymes in the GI tract and poor permeability of insulin through the mucosal barrier. Incorporation into the delivery system of new methods to increase the membrane permeability to macromolecules is essential for attaining a high oral bioavailability that is acceptable in clinical applications (2,5,6).
Recently, CPPs have opened a new horizon for drug delivery of a vast collection of biomolecules that otherwise do not cross the plasma membrane (27,28). CPPs comprise short polycationic peptides based on hydrophobic sequences derived from signal peptides, viral peptides, or other sources (29), and are recognized as promising tools for the intracellular delivery of macromolecular medicines (30–33). Our recent study has shown that the absorption of insulin and other medicinal peptides and proteins across the epithelial mucosal membrane can be improved significantly by the coadministration of new CPPs with biodrug cargos (13–15).
In our recent study of oral peptide/protein delivery systems with high bioavailability based on noncovalent intermolecular interactions with insulin, both l- and d-penetratin seemed to be promising carriers for oral insulin delivery (34). Considering that the apparent insulin permeability varies between different intestinal regions (24,35), there may be a possibility for increasing further insulin oral bioavailability by delivering it to the best site. Thus, we designed this study to investigate whether the site influences the insulin absorption efficiency of the l- and d-forms of CPPs in various intestinal regions and the effects of pretreatment of intestinal membranes with CPPs.
As shown in Fig. 1, insulin absorption following coadministration of l- and d-penetratin differed between the four intestinal regions: duodenum, jejunum, ileum, and colon. The greatest insulin absorption was observed following coadministration with l-penetratin to the ileal segments. The plasma insulin concentration was lower after colonic administration of l-penetratin than after ileal administration (Table II). The greatest insulin absorption from the ileum might reflect differences in proteolytic activity along the intestine. Insulin degradation was much greater in the duodenum and jejunum than in the ileum because the protease content is much higher in the upper small intestine than in the descending small intestine (36). The ranking order for the total different endopeptidase activity in gut luminal fluids and exopeptidase activity in gut mucosal homogenates of rats was jejunum > ileum > colon. The proteolytic activities of trypsin and chymotrypsin in the duodenum are almost twice those in the jejunum and three times those in the ileum (37).
The mucus/glycocalyx layers presumably also provide an enzymatic barrier and/or diffusional barrier. The enzymatic degradation of insulin in the mucus/glycocalyx layers tends to increase toward the upper small intestine in the following order: duodenum > jejunum > ileum (38). By contrast, the colonic coadministration of insulin and penetratin had a smaller effect on increasing insulin absorption, even though the proteolytic activity in the colon may be much lower than that in the small intestine (39). The absorption of insulin from the colon is low, presumably because of the thickness of the mucus layers (40,41) and tightness of tight junctions (42). The mucus layer is a permeation barrier to peptide and protein drug absorption (43,44). It was verified that the diminishment of mucous/glycocalyx layers showed the significant increase of insulin permeability (45). The tight junction is more rigid in the large intestine than in the small intestine, which may contribute to a lower permeability of macromolecular drugs from the large intestine compared with the small intestine (42). As a result of having thicker mucus layers and tightness of mucosal membrane in the colonic region, insulin absorption in the colon may be decreased, even though the insulin degradation with the proteolytic enzyme has a lower extent in the colonic segment than in the small intestine. In this case, the absorption-enhancing effect of d-penetratin for insulin absorption was greater in the colon compared to the small intestine. Thus, it was confirmed that the low intrinsic permeability of insulin and/or the enzymatic degradation in the small intestine was a major barrier for insulin absorption (24).
Additionally, the intestinal pH is an important factor for drug permeability across the intestinal epithelial membrane (46,47). The luminal pH along the GI tract varies between 5 and 8 in rats. It has been shown that the pH varies between 6.6 and 7.1 in the duodenum, it lies in the interval 7.2–8.0 from the jejunum to ileum, while it is in the range of 6.8–7.5 in the colon. Our previous study confirmed that the binding efficiency between a peptide drug and CPP and the enhancement of intestinal peptide drug absorption were affected by altering the pH of the mixture, suggesting that the strength of the electrostatic interaction was affected by a change in the environmental pH which is associated with the enhancement of intestinal absorption by CPP (19). Furthermore, our recent study clearly demonstrated a pH-dependent characteristic of the binding of penetratin to insulin and the efficiency of its enhancement of epithelial insulin permeation through a Caco-2 cell monolayer (48). In the study, a dose-dependent increase in binding responses was observed depending on the l- and d-penetratin concentration at neutral pH (6.0, 7.0, and 8.0). In contrast, the binding of l- and d-penetratin to insulin was particularly decreased at pH 5.0. The permeability of insulin through the epithelial monolayer was enhanced by coincubation with penetratin, especially l-penetratin, at pH 6.0, 7.0, and 8.0; however, the inherent permeability of insulin at pH 5.0 was lower than that at neutral pH.
As mentioned above, l- and d-penetratin stimulate intestinal insulin absorption to different extents. The proteolytic stability of CPPs is a critical requirement for their therapeutic application, and it is essential that they can deliver their cargos to the site of administration effectively without being cleaved by proteases in gut luminal fluids and/or the peptidase of mucus/glycocalyx layers. The differences in the sensitivity to enzymatic degradation between the l- and d-forms of peptides affect the ability of penetratin to increase intestinal insulin absorption. l-Penetratin comprising the l-forms of amino acids is more metabolically unstable in the intestinal lumen than that comprising d-form amino acids (13,15,49). In addition to this proteolytic stability, we should consider another factor. The formation of complexes via electrostatic and/or hydrophobic interactions after mixing insulin and penetratin may protect them from enzymatic degradation. On the other hand, insulin must be dissociated from complexes in the lumen and/or cytoplasm because the bulky size of the complexes hampers their permeation through the basal membrane of the epithelium. The dissociation rate of insulin from the complex is a critical factor in determining the enhancing effect on drug absorption, and the balance of both factors should be considered for evaluating the results (14). In fact, in our in vivo proof of concept of oral insulin study, d-penetratin showed higher pharmacological availability (18.2%) than did l-penetratin (34). The fact is apparently inconsistent with the current study. Despite the lower stability of peptides comprising l-amino acids, l-penetratin caused a greater increase in intestinal insulin absorption than did d-penetratin in this study. However, both experimental conditions are completely different. In the oral absorption study, the physical mixture of insulin and penetratin was exposed to the harsh environment in the oral administration. l-Penetratin could not protect insulin, and both were degraded easily. However, insulin associated with d-penetratin was relatively stable, and with moving along to the small intestine, insulin may be dissociated from d-penetratin by diluting the intestinal fluid. On the other hand, in the in situ study, insulin and penetratin may exist by keeping the intermolecular interaction in a small intestinal segment. The l-penetratin–insulin complex is degraded by intestinal enzymes because of the low resistance of the l-form of the peptide to enzymatic degradation, in contrast to the d-form. In addition, the internalization efficiency of l-penetratin is retained even when its sequence is partially modified, which suggests that fractionated penetratin might retain the ability to internalize a cargo drug (21). Thus, l-penetratin showed a greater increase in intestinal insulin absorption, and this is consistent with our former results obtained by in situ absorption study (13).
Next, we used the CPPs assessed in our previous studies to examine the differences between the abilities of different CPPs to increase insulin absorption (12–15,22,23). The time profiles for plasma insulin concentration after ileal coadministration of insulin with penetratin, R8, or PenetraMax, and the pharmacokinetic parameters are shown in Fig. 2 and Table III, respectively. Among the CPPs, l-penetratin, d-R8, and l- and d-PenetraMax significantly increased ileal insulin absorption. The differences in the chemical structure of these CPP categories account for the differences in their affinity to membrane lipids, membrane insertion capacity, and structural transitions upon binding with negatively charged moieties in the mucus layer of the epithelial membrane (50). Penetratin is an amphipathic CPP that traverses pure lipid model membranes used in drug interaction studies. By contrast, cationic CPPs such as R8 have a lower translocation ability under these conditions. PenetraMax is a rearranged and sequence-modified penetratin analog developed from an in silico design based on the molecular orbital method with a self-organizing map classification (22).
The number of arginine residues in l-R8 is important to the internalization efficiency, and the effects of this CPP decrease markedly with a decreasing number of arginines (12). By contrast, the internalization efficiency of penetratin is retained even when its sequence is partially modified as described above, which suggests that fractionated penetratin displays an internalization ability (21,22). The superior enhancing effect of PenetraMax on intestinal insulin absorption may be attributed to the change in the positions of the aromatic amino acid tryptophan (Table I) (21,22,49). This would allow a high degree of conformational flexibility of the interacting moieties, thus stabilizing the conformation of the peptide at the water–lipid interface and facilitating the insertion of PenetraMax into the lipid bilayer (51,52). In the case of PenetraMax, there was no difference between the l- and d-forms in increasing insulin absorption. This tendency is consistent with their nasal interferon absorption-enhancing effect (data not shown). To clarify the mechanism, an intermolecular interaction study of insulin and PenetraMax under the same experimental in situ ileal absorption conditions shows that the bound concentrations of penetratin and PenetraMax were 66.2 and 91.9% of the total concentration, respectively (22). Whatever the case, the greater capacity of PenetraMax to interact with insulin than the parent penetratin might be involved in its enhancing effect on ileal absorption of insulin, especially in our approach using physical mixtures of the drug and CPPs rather than their conjugates (19,20,22).
We found that l- and d-PenetraMax were the most effective absorption enhancers for insulin among the CPPs assessed in the present study. However, despite this ability of PenetraMax, only a slightly increased insulin absorption was observed after administration of insulin into the ileal loop that had been pretreated with l- and d-PenetraMax for 3 h (Figs. 4 and 5). This result suggests two important aspects. First is that the intermolecular interaction between insulin and a CPP is a key factor governing the absorption-enhancing effect of CPPs because only a small effect was observed following insulin administration after removal of PenetraMax. Second is that PenetraMax has no irreversible effect on the mucosal structure, which means that CPPs do not induce permanent functional change to the intestinal membrane.
As mentioned above, unlike penetratin and R8, the difference between the enhancement efficiency of l- and d-PenetraMax is not significant (Table III). In addition to the enhancing effect of CPPs on insulin permeation across the epithelial membranes (53,54), our current results suggest that the protective effect of CPPs on insulin degradation in the intestinal lumen is involved in the increase in insulin absorption (34). Hence, we evaluated insulin degradation in rat intestinal enzymatic fluid in the presence of the l- and d-forms of penetratin, R8, and PenetraMax. As expected, insulin degradation in the intestinal enzymatic fluid was faster in the presence of the l-forms of penetratin and R8 than in the presence of the d-forms (Fig. 5a, b and Table IV), reflecting the differences in their absorption-enhancing effects. Similarly, the stabilizing effect for insulin was significantly stronger for d-PenetraMax than for l-PenetraMax (Fig. 5c and Table IV), but this difference did not match the differences in their absorption-enhancing efficiencies. At present, we cannot identify the mechanisms responsible for the different pattern of absorption enhancement by PenetraMax, but the permeability of insulin–PenetraMax complexes through the epithelium may be related to the similar ability of l- and d-PenetraMax to improve ileal insulin absorption. The increase in permeability caused by the sequence-modified peptide analog may contribute to the high bioavailability of this drug (55). The stabilizing effect of l- and d-PenetraMax on insulin degradation appears to be a separate means of increasing insulin absorption that works synergistically with the membrane-penetrating effect. However, the mechanism underlying the membrane-penetrating effect of PenetraMax needs further detailed studies.
CONCLUSIONS
In this study, we demonstrated that the amphipathic CPP penetratin has a region-dependent enhancement effect on intestinal insulin absorption. In addition to the improved permeation through epithelial cells, the ability of CPPs to protect insulin against enzymatic degradation appears to contribute to the increase in intestinal insulin absorption. The chirality of the constituent amino acids of CPPs affects the absorption-enhancing efficiency and stability of insulin in the intestinal enzymatic fluid. In the case of l- and d-forms of penetratin, the most effective target delivery site in terms of increasing oral bioavailability would be the ileum and the colon, respectively. Among the CPPs examined in this study, PenetraMax was the most effective in increasing the absorption of insulin from the ileum; the l- and d-forms had similar effectiveness. PenetraMax had no irreversible effects on the ileal mucosal structure. The current investigation provides a foundation for the clinical realization of an effective oral insulin delivery system using CPPs as potential enhancers of insulin bioavailability.
References
Mustata G, Dinh SM. Approaches to oral drug delivery for challenging molecules. Crit Rev Ther Drug Carrier Syst. 2006;23:111–1135.
Morishita M, Peppas NA. Is the oral route possible for peptide and protein drug delivery? Drug Discov Today. 2006;11:905–10.
Gerstein HC, Miller ME, Byington RP, Goff DC, Bigger JT, Buse JB, et al. Effects of intensive glucose lowering in type 2 diabetes. New Engl J Med. 2008;358:2545–59.
Iyer H, Khedkar A, Verma M. Oral insulin—a review of current status. Diabetes Obes Metab. 2010;12:179–85.
Goldberg M, Gomez-Orellana I. Challenges for the oral delivery of macromolecules. Nat Rev Drug Discov. 2003;2:289–95.
Khafagy ES, Morishita M, Onuki Y, Takayama K. Current challenges in non-invasive insulin delivery systems: a comparative review. Adv Drug Deliv Rev. 2007;59:1521–46.
Lin YH, Sonaje K, Lin KM, Juang JH, Mi FL, Yang HW, et al. Multi-ion-crosslinked nanoparticles with pH-responsive characteristics for oral delivery of protein drugs. J Control Release. 2008;132:141–9.
Woitiski CB, Veiga F, Ribeiro A, Neufeld R. Design for optimization of nanoparticles integrating biomaterials for orally dosed insulin. Eur J Pharm Biopharm. 2009;73:25–33.
Yamagata T, Morishita M, Kavimandan NJ, Nakamura K, Fukuoka Y, Takayama K, et al. Characterization of insulin protection properties of complexation hydrogels in gastric and intestinal enzyme fluids. J Control Release. 2006;112:343–9.
Sonaje K, Chen YJ, Chen HL, Wey SP, Juang JH, Nguyen HN, et al. Enteric-coated capsules filled with freeze-dried chitosan/poly(gamma-glutamic acid) nanoparticles for oral insulin delivery. Biomaterials. 2010;31:3384–94.
Su FY, Lin KJ, Sonaje K, Wey SP, Yen TC, Ho YC, et al. Protease inhibition and absorption enhancement by functional nanoparticles for effective oral insulin delivery. Biomaterials. 2012;33:2801–11.
Morishita M, Kamei N, Ehara J, Isowa K, Takayama K. A novel approach using functional peptides for efficient intestinal absorption. J Control Release. 2007;118:177–84.
Kamei N, Morishita M, Eda Y, Ida N, Nishio R, Takayama K. Usefulness of cell-penetrating peptides to improve intestinal insulin absorption. J Control Release. 2008;132:21–5.
Khafagy ES, Morishita M, Isowa K, Imai J, Takayama K. Effect of cell-penetrating peptides on the nasal absorption of insulin. J Control Release. 2009;133:103–8.
Khafagy ES, Morishita M, Kamei N, Eda Y, Ikeno Y, Takayama K. Efficiency of cell-penetrating peptides on the nasal and intestinal absorption of therapeutic peptides and proteins. Int J Pharm. 2009;381:49–55.
Derossi D, Joliot AH, Chassaing G, Prochiantz A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem. 1994;269:10444–50.
Derossi D, Calvet S, Trembleau A, Brunissen A, Chassaing G, Prochiantz A. Cell internalization of the third helix of the Antennapedia homeodomain is receptor-independent. J Biol Chem. 1996;271:18188–93.
Drin G, Cottin S, Blanc E, Rees AR, Temsamani J. Studies on the internalization mechanism of cationic cell-penetrating peptides. J Biol Chem. 2003;278:31192–201.
Kamei N, Morishita M, Takayama K. Importance of intermolecular interaction on the improvement of intestinal therapeutic peptide/protein absorption using cell-penetrating peptides. J Control Release. 2009;136:179–86.
Khafagy ES, Morishita M, Takayama K. The role of intermolecular interactions with penetratin and its analogue on the enhancement of absorption of nasal therapeutic peptides. Int J Pharm. 2010;388:209–12.
Khafagy ES, Morishita M, Ida N, Nishio R, Isowa K, Takayama K. Structural requirements of penetratin absorption enhancement efficiency for insulin delivery. J Control Release. 2010;143:302–10.
Kamei N, Kikuchi S, Takeda-Morishita M, Terasawa Y, Yasuda A, Yamamoto S, et al. Determination of the optimal cell-penetrating peptide sequence for intestinal insulin delivery based on molecular orbital analysis with self-organizing maps. J Pharm Sci. 2013;102:469–79.
Khafagy ES, Kamei N, Nielsen EJB, Nishio R, Takeda-Morishita M. One-month subchronic toxicity study of cell-penetrating peptides for insulin nasal delivery in rats. Eur J Pharm Biopharm. 2013;85:736–43.
Morishita M, Morishita I, Takayama K, Machida Y, Nagai T. Site-dependent effect of aprotinin, sodium caprate, Na2EDTA and sodium glycocholate on intestinal absorption of insulin. Biol Pharm Bull. 1993;16:68–72.
Pantzar N, Weström BR, Luts A, Lundin S. Regional small-intestinal permeability in vitro to different-sized dextrans and proteins in the rat. Scand J Gastroenterol. 1993;28:205–11.
Asada H, Douen T, Mizokoshi Y, Fujita T, Murakami M, Yamamoto A, et al. Stability of acyl derivatives of insulin in the small intestine: relative importance of insulin association characteristics in aqueous solution. Pharm Res. 1994;11:1115–20.
Foged C, Nielsen HM. Cell-penetrating peptides for drug delivery across membrane barriers. Expert Opin Drug Deliv. 2008;5:105–17.
Patel LN, Zaro JL, Shen WC. Cell penetrating peptides: intracellular pathways and pharmaceutical perspectives. Pharm Res. 2007;24:1977–92.
Hawiger J. Noninvasive intracellular delivery of functional peptides and proteins. Curr Opin Chem Biol. 1999;3:89–94.
Gupta B, Levchenko TS, Torchilin VP. Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides. Adv Drug Deliv Rev. 2005;57:637–51.
Foerg C, Merkle HP. On the biomedical promise of cell penetrating peptides: limits versus prospects. J Pharm Sci. 2008;97:144–62.
Khafagy ES, Morishita M. Oral biodrug delivery using cell-penetrating peptide. Adv Drug Deliv Rev. 2012;64:531–9.
Jones AT, Sayers EJ. Cell entry of cell penetrating peptides: tales of tails wagging dogs. J Control Release. 2012;161:582–91.
Nielsen EJB, Yoshida S, Kamei N, Iwamae R, Khafagy ES, Olsen J, et al. In vivo proof of concept of oral insulin delivery based on a co-administration strategy with the cell-penetrating peptide penetratin. J Control Release. 2014;189:19–24.
Schilling RJ, Mitra K. Intestinal mucosal transport of insulin. Int J Pharm. 1990;62:53–64.
Tozaki H, Odoriba T, Iseki T, Taniguchi T, Fujita T, Murakami M, et al. Use of protease inhibitors to improve calcitonin absorption from the small and large intestine in rats. J Pharm Pharmacol. 1998;50:913–20.
Layer P, Jansen JB, Cherian L, Lamers CB, Goebell H. Feedback regulation of human pancreatic secretion. Effects of protease inhibition on duodenal delivery and small intestinal transit of pancreatic enzymes. Gastroenterology. 1990;98:1311–9.
Aoki Y, Morishita M, Asai K, Akikusa B, Hosoda S, Takayama K. Region-dependent role of the mucous/glycocalyx layers in insulin permeation across rat small intestinal membrane. Pharm Res. 2005;22:1854–62.
Bai JP. Subcellular distribution of proteolytic activities degrading bioactive peptides and analogues in the rat small intestinal and colonic enterocytes. J Pharm Pharmacol. 1994;46:671–5.
Atuma C, Strugala V, Allen A, Holm L. The adherent gastrointestinal mucus gel layer: thickness and physical state in vivo. Am J Physiol Gastrointest Liver Physiol. 2001;280:G922–9.
Ermund A, Schütte A, Johansson ME, Gustafsson JK, Hansson GC. Studies of mucus in mouse stomach, small intestine, and colon. I. Gastrointestinal mucus layers have different properties depending on location as well as over the Peyer’s patches. Am J Physiol Gastrointest Liver Physiol. 2013;305:G341–7.
Tanaka Y, Taki Y, Sakane T, Nadai T, Sezaki H, Yamashita S. Characterization of drug transport through tight-junctional pathway in Caco-2 monolayer: comparison with isolated rat jejunum and colon. Pharm Res. 1995;12:523–8.
Winne D, Verheyen W. Diffusion coefficient in native mucus gel of rat small intestine. J Pharm Pharmacol. 1990;42:517–9.
Larhed AW, Artursson P, Björk E. The influence of intestinal mucus components on the diffusion of drugs. Pharm Res. 1998;15:66–71.
Morishita M, Aoki Y, Sakagami M, Nagai T, Takayama K. In situ ileal absorption of insulin in rats: effects of hyaluronidase pretreatment diminishing the mucous/glycocalyx layers. Pharm Res. 2004;21:309–16.
Ward FW, Coates ME. Gastrointestinal pH measurement in rats: influence of the microbial flora, diet and fasting. Lab Anim. 1987;21:216–22.
McConnell EL, Basit AW, Murdan S. Measurements of rat and mouse gastrointestinal pH, fluid and lymphoid tissue, and implications for in-vivo experiments. J Pharm Pharmacol. 2008;60:63–70.
Kamei N, Aoyama Y, Khafagy ES, Henmi M, Takeda-Morishita M. Effect of different intestinal conditions on the intermolecular interaction between insulin and cell-penetrating peptide penetratin and on its contribution to stimulation of permeation through intestinal epithelium. Eur J Pharm Biopharm. 2015;94:42–51.
Pappenheimer JR, Dahl CE, Karnovsky ML, Maggio JE. Intestinal absorption and excretion of octapeptides composed of D amino acids. Proc Natl Acad Sci U S A. 1994;91:1942–5.
Ziegler A, Seelig J. High affinity of the cell-penetrating peptide HIV-1 Tat-PTD for DNA. Biochemistry. 2007;46:8138–45.
Lensink MF, Christiaens B, Vandekerckhove J, Prochiantz A, Rosseneu M. Penetratin-membrane association: W48/R52/W56 shield the peptide from the aqueous phase. Biophys J. 2005;88:939–52.
Polyansky AA, Volynsky PE, Arseniev AS, Efremov RG. Adaptation of a membrane-active peptide to heterogeneous environment. I. Structural plasticity of the peptide. J Phys Chem B. 2009;113:1107–19.
Kamei N, Onuki Y, Takayama K, Takeda-Morishita M. Mechanistic study of the uptake/permeation of cell-penetrating peptides across a caco-2 monolayer and their stimulatory effect on epithelial insulin transport. J Pharm Sci. 2013;102:3998–4008.
Kamei N, Morishita M, Ehara J, Takayama K. Permeation characteristics of oligoarginine through intestinal epithelium and its usefulness for intestinal peptide drug delivery. J Control Release. 2008;131:94–9.
Ziegler A. Thermodynamic studies and binding mechanisms of cell-penetrating peptides with lipids and glycosaminoglycans. Adv Drug Deliv Rev. 2008;60:580–97.
Acknowledgments
This study was supported in part by JSPS KAKENHI Grant Number 23590056. This work was also supported in part by the Japan Student Services Organization.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Khafagy, ES., Iwamae, R., Kamei, N. et al. Region-Dependent Role of Cell-Penetrating Peptides in Insulin Absorption Across the Rat Small Intestinal Membrane. AAPS J 17, 1427–1437 (2015). https://doi.org/10.1208/s12248-015-9804-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12248-015-9804-y