
Abstract
The term ‘biosimilar’ refers to an alternative similar version of an off-patent innovative originator biotechnology product (the ‘reference product’). Several biosimilars have been approved in Europe, and a number of top-selling biological medicines have lost, or will lose, patent protection over the next 5 years. We look at the experience in Europe so far. The USA has finally implemented a regulatory route for biosimilar approval. We recommend that European and US governments and payers take a strategic approach to get value for money from the use of biosimilars by (1) supporting and incentivising generation of high-quality comprehensive outcomes data on the effectiveness and safety of biosimilars and originator products; and (2) ensuring that incentives are in place for budget holders to benefit from price competition. This may create greater willingness on the part of budget holders and clinicians to use biosimilar and originator products with comparable outcomes interchangeably, and may drive down prices. Other options, such as direct price cuts for originator products or substitution rules without outcomes data, are likely to discourage biosimilar entry. With such approaches, governments may achieve a one-off cut in originator prices but may put at risk the creation of a more competitive market that would, in time, produce much greater savings. It was the creation of competitive markets for chemical generic drugs—notably, in the USA, the UK and Germany—rather than price control, that enabled payers to achieve the high discounts now taken for granted.
Similar content being viewed by others

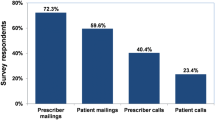

Payers and decision makers need to think medium/long term if they want to achieve significant, sustained savings from use of biosimilars. |
High-quality comprehensive outcomes data on the effectiveness and safety of biosimilars and originator products are required to ensure that they can be used interchangeably. |
Incentives are also needed for budget holders to benefit from using biosimilars. |
Over time, in some therapy areas, outcomes data may lead to discounts approaching chemical generic levels and support the introduction of substitution rules. |
Initiatives focusing on short-term savings—such as price cuts for originators, reference pricing or substitutability rules (without the outcomes data mentioned above)—are likely to put at risk the creation of a more competitive market that would, in time, produce much greater savings. |
1 Introduction
Biotherapeutic or biological medicines have active ingredients derived from proteins (such as growth hormone, insulin and antibodies) and other substances produced by living organisms (such as cells, viruses and bacteria). They are larger and more complex than chemically synthesised medicines, are therefore harder to manufacture and are usually administered as an infusion. There has been a rapid worldwide increase in the number of biological medicines receiving regulatory approval.
The term ‘biosimilar’ refers to an off-patent version of an innovative ‘reference’ biologic. The European Medicines Agency (EMA) states that “a similar biological or biosimilar medicine is a biological medicine that is similar to another biological medicine that has already been authorised for use” [1] and that “Biosimilars are not the same as generics, which have simpler chemical structures and are considered to be identical to their reference medicines” [2]. In line with the EMA, the US Food and Drug Administration (FDA) states that “A biosimilar product is a biological product that is approved based on a showing that it is highly similar to an FDA-approved biological product, the reference product, and has no clinically meaningful differences in terms of safety and effectiveness from the reference product” [3]. A number of biosimilars have been approved already by the EMA: erythropoietins (EPOs), granulocyte colony-stimulating factors (GCSFs), growth hormones and, more recently, monoclonal antibodies (mAbs). The GCSF Zarxio (filgrastim-sndz) was the first biosimilar product approved by the FDA, in March 2015, and was available for launch from September 2015.
We set out proposals building on previous analysis [4] to ensure that European and US third-party payers can secure value from biosimilars, as many top-selling biological medicines have lost, or will lose, patent protection over the next 5 years. Before doing so, we outline (1) the EU regulatory process; (2) economic characteristics of biosimilars and evidence from Europe; (3) differences from chemical generics; and (4) developments in the US market, where biosimilar regulatory pathways have lagged behind those in Europe.
2 Regulatory Processes in Europe
EMA biosimilar pathway guidelines were first published in 2005, followed by a number of product-specific guidelines. An extensive comparability exercise, including clinical work, is required to demonstrate quality, safety and efficacy similar to those of the reference product.
Assessment of substitution and interchangeability with the originator product is not part of the EMA’s scientific evaluation [2] but is a Member State competency. The EMA states that patients should address questions related to switching from one biological medicine to another to their doctor and pharmacist. Substitution of biologics (including between an innovator and a biosimilar) by a pharmacist without the permission of the prescribing doctor either is not allowed or is advised against in the vast majority of EU countries (including Italy, Spain, the UK, the Netherlands, Sweden and Germany). This includes countries that do allow chemical generic substitution.
3 Competition in Europe’s Biosimilars Market
Initial research on biosimilar entry [5–7] suggested that competition between a reference product and biosimilar(s) would resemble brand-to-brand competition (focusing on price, quality and promotion) rather than generic-to-brand competition. Experience to date confirms this hypothesis. Competition is, however, still evolving.
Evidence suggests that it is difficult to generalise across either therapy areas or countries in Europe, for four reasons. First, clinician willingness and receptiveness to use biosimilars across therapy areas differ, resulting in higher biosimilar penetration for GCSFs and EPOs than for growth hormones [4, 8]. Second, the scope for biosimilar competition is lessened for markets with high price competition pre-biosimilar entry. Third, biosimilar penetration differs across countries according to the incentives implemented to encourage usage. Germany, Sweden and the UK—all successful chemical generic markets—have relatively high biosimilar penetration. Fourth, some biosimilars are administered by physicians in clinics and hospitals, while others are self-administered or are administered by home health providers and dispensed through specialty retail pharmacies. Incentives to promote biosimilar use differ among channels. Regulations and incentives to promote biosimilar uptake primarily impact at the hospital and physician levels. Retail pharmacists have little incentive to promote biosimilar uptake [9].
Analyses shows disparities of biosimilar penetration across Europe [10–12]. Germany, with strong incentives to encourage biosimilar uptake, has the highest penetration rates in the EPO market, where volume sales of the originator product were matched by those of biosimilars in 2009 [13] in spite of the originator cutting its price. By the end of 2011, biosimilars accounted for more than 60 % in volume terms [8]. EPOs are mainly dispensed in retail pharmacists in Germany, and substitution rules (as well as prescription incentives; see below) have favoured EPO biosimilar uptake [12]. For GCSFs, however, the originator’s price has remained stable, and the biosimilars’ market share accounted for less than 50 % by the end of 2011. Omnitrope, a biosimilar growth hormone, has had much lower uptake in spite of a price discount of around 30 % [13]. It seems that clinicians have had concerns about efficacy and safety in the paediatric population it serves [14]. It has been argued that the German base of many biosimilar companies has also helped the uptake in Germany [8, 12]. Penetration of biosimilars in Germany has occurred in spite of reference pricing (RP), which we would expect to discourage biosimilar entry and use by pulling down the price of the originator. There are, however, four important offsetting factors. First, the RP calculation generates higher prices to incentivise further market entry when there are fewer generic competitors [15, 16]. Second, drugs are generally exempt from co-payment if they are priced at 30 % or more below the RP, increasing the attractiveness to clinicians of prescribing them [17]. Third, payers and manufacturers can negotiate confidential rebates, which are not reflected in the list price used for referencing. As part of these agreements, payers often also exempt patients from co-payment. Fourth, a quota system, by which physicians have to prescribe a certain percentage of biosimilars, has encouraged prescribing [11, 12, 16].
In Sweden, the biosimilar penetration rates for both EPOs and GCSFs have exceeded 60 %. In the UK, the rates are 10 and 80 %, respectively. The low UK biosimilar EPO uptake reflects high discounting by competing brands prior to biosimilar entry [19]. The penetration rates for GCSF biosimilars (filgrastim—reference product Neupogen; a number of biosimilars exist, including Zarzio, Tevagrastim, Ratiograstim and Nivestim) in Italy and France have approached 45 and 60 %, respectively, with EPO biosimilar use being around 15 % in both countries [19]. Greater acceptance of GCSF biosimilars in these two countries may be the consequence of medical considerations and/or reimbursement policies [8, 20]. The Italian Agency for Medicines (AIFA) has approved eight of 12 EMA-approved biosimilars; Italian Regional Health Authorities have facilitated biosimilar entry and price competition with tenders [21]. However, Spain also uses tenders but has minimal biosimilar use [22].
Austria’s application of its generic pricing policy to biosimilars represents an unusual, somewhat simplistic, biosimilar pricing policy in Europe. The first biosimilar that is launched must be priced at 52 % of the reference drug, the second at 44 % and the third at 40 %. The reference drug and the other two biosimilars must be priced at 40 % of the originator’s original price when the third biosimilar enters. This policy has discouraged biosimilar competition, and no third biosimilar has been launched in Austria [18].
Entry by second-generation improvements on ‘reference products’ before biosimilar entry, for both GCSFs and EPOs, cannibalised sales of the first-generation products across European countries [8]. These second-generation products require substantially fewer infusions over a course of treatment, with potential benefits to patients and lower administration costs [8]. As with chemical generics, successful second-generation products reduce the potential for cost savings by reducing the market share of the first-generation reference product subject to biosimilar competition.
Two infliximab biosimilars (Remsima and Inflectra; reference product Remicade) were approved by the EMA in 2013 [4]. Norway offers an interesting case study, for two reasons: (1) use of tenders; and (2) promotion of outcomes studies exploring the impact of switching. Biosimilar penetration in Norway for GCSFs and EPOs is among the highest in Europe. In January 2014, Orion Pharma (which distributes Celltrion’s Remsima in Scandinavia and Estonia) secured the first position for its biosimilar product Remsima in the tender for national supply of infliximab by offering a 39 % discount against Remicade [23, 24]. However, in March 2015, Orion proposed a 72 % price reduction for Remsima [23]. This level of discount has stunned industry observers, as much lower discounts were expected [23]. To promote a substitution culture, the Norwegian Medicines Agency is funding a clinical study (the NOR-SWITCH study) [25] to explore Remicade and Remsima/Inflectra’s interchangeability. The trial is still recruiting patients, and the results are expected in April 2016. In July 2015, the Dutch authorities began funding a similar trial (BIO-SWITCH [26, 27]) to study the effects on efficacy, safety and immunogenicity of switching treatment from Remicade to an infliximab biosimilar in patients with rheumatoid arthritis, spondyloarthritis or psoriatic arthritis. The results are expected in April 2017.
4 Economics of Biosimilars: Differences from Chemical Generics
Economic theory suggests five reasons not to expect similar levels of price discounting for biosimilars in comparison with chemical generics. First, biosimilar development costs are considerably higher, as biosimilars require pre-clinical and clinical studies. Second, biologic manufacturing costs are higher. Third, manufacturers need to communicate with prescribers as well as pharmacists, as prescribing is done by brand name. Originators have established relationships with prescribers, key opinion leaders and patients, based on services, clinical development and data. Biosimilar-only manufacturers cannot replicate these without substantial investment [28]. Fourth, physician (and pharmacist) concerns about comparability may need to be addressed in post-launch studies [6, 29, 30], raising costs and reducing adoption rates relative to those of chemical generics. We can expect lower sales initially because of concerns on the part of some physicians regarding the degree of substitutability between the reference product and its biosimilar(s). Fifth, because of all of the above, we can expect fewer biosimilar entrants and consequently less intense biosimilar price competition [31–33].
These factors (development costs; manufacturing costs; potential prescriber concerns, raising entry hurdles; the need for post-launch data; and the degree of competition) help to explain why price differentials including discounts (to date) between biosimilars and their reference products in Europe can typically be less than 30 % [8, 10, 11] as compared with 80 % plus discounts for chemical generic drugs—again, with the caveat that there are differences across therapy areas and countries, and noting the recent exception of infliximab’s biosimilar discount in Norway.
Prices need to be high enough for long enough to reward biosimilar manufacturers if a competitive market is to exist over time. Given these factors, the long-term equilibrium price for a sustainable biosimilars market should be somewhere between the long-run equilibrium for a chemical generic market—albeit with higher manufacturing costs impacting the level of pricing—and a monopolistic competition-type situation (with a large number of sellers, and with entry and exit costs being zero) in which manufacturers have to maintain a higher cost base in order to invest in their product, which is how biosimilars were first modelled [5, 34]. In practice, discounts against reference products could increase to generic levels over time if outcomes studies support interchangeability and/or substitution [6].
Evidence from chemical generics shows that price regulation for entry generally appears to be associated with reduced incentives and limited diffusion after entry [35, 36]. On the other hand, effective demand-side incentives—including physician budgets linked to incentives, generic substitution (possibly linked to incentives) and generic prescribing incentives—encourage generic usage [37, 38]. We can expect similar effects on biosimilar use from price regulation (negative) and demand incentives (positive).
5 The US Market: Projected Cost Savings and Recent Developments
The US lags behind Europe in developing a clearly defined regulatory pathway for biosimilars. In March 2010, the US Congress established an abbreviated approval pathway for biosimilars (under the 2009 Biologics Price Competition and Innovation Act [BPCIA]). Recently, the FDA has implemented a regulatory route for biosimilar approval, albeit that uncertainties remain as to the extent of the evidence required by the FDA. This will affect entry costs and thus the nature of biosimilar competition.
The FDA is devising a process to establish similarity along EMA lines. The legislation, however, also requires it to provide guidance on “a higher standard of similarity to a reference product—interchangeability—reflecting an FDA assessment that pharmacists can make substitutions between biologics without the prescribers’ intervention” [31]. Achieving a favourable FDA interchangeability opinion (note that in the USA, this refers to substitutability at the pharmacy level) might require manufacturers to undertake additional switching clinical trials and other studies [20, 39]. The benefits would be pharmacist substitution and reduced need for post-launch outcomes research.
In July 2015, the US Court of Appeals for the Federal Circuit concluded the Amgen versus Sandoz Zarxio case. At issue were two BPCIA provisions: (1) that the applicant “shall” provide to the reference product sponsor a copy of its application and also manufacturing information; (2) that the biosimilar applicant will provide 180 days’ notice to the reference product sponsor that it intends to enter the marketplace. The Court held the contemplated information exchange as being non-mandatory. However, if an applicant filing an abbreviated Biologics License Application (BLA) fails to participate in that information exchange, it must wait for FDA approval of its biosimilar product prior to providing 180 days’ advance notice to the reference product sponsor that it intends to begin commercial marketing [40]. Sandoz (Zarxio’s applicant) did not provide the information to Amgen (the manufacturer of the reference product, Neupogen). Given that Zarxio obtained FDA approval on 5 March 2015, it could be marketed from 2 September 2015.
Estimates of savings from biosimilars over a 10-year framework in the USA have ranged between $25 billion and $100 billion [41–43]. A review of estimates for both the EU and the USA has been set out by Rovira et al. [13]. The differences in savings arise from different assumptions for two key parameters: penetration rates and price discounts. More aggressive assumptions on biosimilar uptake and price discounts yield higher potential cost savings. Two additional assumptions affect forecast savings: (1) baseline spending on biologics; and (2) the timing of US regulatory pathway implementation.
Grabowski et al. [8] highlighted how uncertainties around the regulatory pathway might reduce projected cost savings in the short/medium term. Only favourable experiences with biosimilars over the next few years, and regulatory clarity, will enable biosimilars to produce substantial cost savings.
They also noted [8] that delays in establishing a biosimilar regulatory route have led some companies to consider filing using the BLA route, i.e. as a new biologic drug. This route is also available for ‘biobetters’—taken to mean biologics from the same molecule but with modifications giving superiority (e.g. in the delivery mechanism).
6 Recommendations
Our recommendations distinguish between pricing and reimbursement systems that use health technology assessment (HTA)—and, in particular, cost-effectiveness analysis—and those that do not. There are also differences between the EU and US markets [19].
For those systems not relying systematically on HTA, there are five levers for intervention by government in biosimilars markets:
-
1.
Substitutability rules, permitting pharmacists to substitute one biosimilar for another.
-
2.
Direct price intervention, pushing down originator product prices in the form of:
-
(a)
Originator inclusion in RP systems with biosimilars.
-
(b)
A post-patent expiry price cut imposed on the innovator and/or a cut imposed on a biosimilar entrant.
-
(a)
-
3.
Tendering procedures to facilitate biosimilar competition and price reductions.
-
4.
Incentives for budget holders to use lower-cost products when these are safe and effective, and so provide better value for money.
-
5.
Market support (e.g. investing in infrastructure for outcomes monitoring and facilitating pharmacovigilance work, collecting real-world evidence), creating greater willingness on the part of budget holders and clinicians to seek value for money by using biosimilars and originator products interchangeably.
With regard to lever 1, governments are understandably reluctant to implement automatic substitution. An exception is France, which has introduced legislation (on 1 January 2014) allowing substitution of biosimilars but only for a patient beginning a course of treatment; substitution cannot be made part-way through a course. It cannot be done if the prescriber has written “non-substitutable” on the form. Any replacement product would have to be included in ‘similar biologic’ groups, which are still to be drawn up [12]. We recommend that substitution in a therapy area be implemented at a much later stage when strong biosimilar real-world evidence is available in that therapy area.
Lever 2 (direct price intervention) is counterproductive. RP, for example, assumes a degree of interchangeability not initially likely to be reflected in clinicians’ willingness to switch products. More fundamentally, it may preclude collection of real-world evidence and discourage biosimilar entry by reducing potential biosimilar revenues, such that biosimilar manufacturers might not expect to recover one or both of their development costs and costs of collecting post-launch outcomes data. In our view, it is not possible to ‘jump start’ a biosimilars market by forcing down prices or by imposing substitutability. Governments may achieve a one-off cut, but they put at risk the creation of more competitive markets that would, in time, produce much greater savings. We note the use of RP in Germany, but we conclude above that the particular features of the calculation mechanism, interaction with co-payments, confidential discounts and incentives for physicians negate its adverse effects on biosimilar entry. The experience with generics elsewhere highlights how strict price regulation can hinder competition [35, 36].
Lever 3 (biosimilar tendering) is being used in Norway, Italy and Spain. However, the evidence from their use is mixed. The Italian experience suggests that tenders can drive down prices and generate savings, depending on the number of manufacturers participating in the tender [21]. ‘Winner takes all’ tenders also pose another barrier to entry; the risk of not winning the tender and thus not gaining any market share can discourage entry. Another potential problem with tenders is the risk of shortages. If there is a supply shortage from the tender winner, substitution is expensive.
Given the relatively efficient way in which biosimilars markets are likely to evolve, we would recommend use of levers 4 and 5. Financial incentives to use biosimilars are important and account for the progress of biosimilars in those European markets with successful chemical generic markets. Germany has the most favourable incentives and the most successful biosimilar penetration rates. Grabowski et al. [8] noted that incentives for US biosimilar use were addressed in the Affordable Care Act by giving clinicians the same Medicare reimbursement amount as the originator. Yet clinician concerns about the exact nature of ‘similar’ outcomes have to be addressed.
We regard lever 5 as key for governments taking a more strategic approach. We recommend supporting and incentivising generation and use of high-quality comprehensive outcomes data on the effectiveness and safety of biosimilars and originator products. In European markets, clinicians’ receptiveness and willingness to use biosimilars determine biosimilar adoption rates. Outcomes studies can reinforce prescribers’ confidence in biosimilars. They could also explore the value for money of second-generation biotech products that are competing with the first-generation originator and biosimilar products. Government/payer/industry collaboration to determine how to generate these outcomes data and value-for-money data would be helpful. The results should be made public once they are available. Supporting market evolution and sustainability in this way will secure a path towards maximising price competition over time, enabling payers and patients to gain substantial savings from biologic patent expiry. The NOR-SWITCH and BIO-SWITCH studies are important examples of the use of this approach.
We recommend that HTA-driven systems not treat biosimilars as generics, which are usually automatically excluded from new drug appraisals. Two factors should determine whether biosimilars are subject to HTA:
-
1.
Has the reference product been appraised?
-
2.
Is the reference product the standard of care?
If the reference product has been appraised and is the standard of care, an HTA process is not needed for biosimilars at launch. If there is an appraisal, a cost-minimisation analysis would suffice, as there is no more evidence on outcomes than those available to the regulator.
If the reference product was not appraised or was appraised but is not the standard of care, because it was originally rejected or restricted, then the rationale for appraising the biosimilar is stronger. Under this scenario, cost–utility analysis is more appropriate, and the comparator should be the standard of care and not the reference product.
7 Conclusions
Evidence on the nature of competition between biosimilars and their reference products in Europe confirms the earlier hypothesis that such competition would not resemble the aggressive price competition associated with chemical generics. There are, however, important differences across therapy areas and countries.
We believe that levers 4 and 5 are the right direction of travel for governments and payers to maximise long-term savings from biosimilars. Prescribers need confidence in outcomes, and they and/or the health system need to benefit financially from using biosimilars. The Norwegian and Dutch initiatives to collect real-world evidence is a move in the right direction. The results from such studies will impact use positively if the results are favourable. Such studies will become even more important if payers are to reap the benefits from the next wave of biosimilars—notably, mAbs—where concerns over outcomes and safety may be greater.
References
European Medicines Agency [EMA]. Biosimilar medicines. London: EMA; 2013. http://www.ema.europa.eu/ema/index.jsp?curl=pages/special_topics/document_listing/document_listing_000318.jsp&mid=WC0b01ac0580281bf0. Accessed 28 April 2015.
European Medicines Agency [EMA]. Questions and answers on biosimilar medicines (similar biological medicinal products) [document reference EMA/837805/2011]. London: EMA; 2012. http://www.ema.europa.eu/docs/en_GB/document_library/Medicine_QA/2009/12/WC500020062.pdf. Accessed 8 Jan 2016.
US Food and Drug Administration [FDA]. Biosimilars. Silver Spring: FDA; 2015. http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/default.htm. Accessed 14 Sept 2015.
Mestre-Ferrandiz J, Towse A. What is the role of HTA for biosimilars? [briefing no. 54]. London: Office of Health Economics; 2014. https://www.ohe.org/publications/what-role-hta-biosimilars. Accessed 8 Jan 2016.
Grabowski HG, Ridley DB, Schulman KA. Entry and competition in generic biologicals. Manag Decis Econ. 2007;28(4–5):439–51.
Chauhan D, Towse A, Mestre-Ferrandiz J. The market for biosimilars: evolution and policy options [briefing no. 45]. London: Office of Health Economics; 2008. https://www.ohe.org/publications/market-biosimilars-evolution-and-policy-options. Accessed 8 Jan 2016.
Mattison N, Mestre-Ferrandiz J, Towse A, editors. Biosimilars: how much entry and price competition will result? London: Office of Health Economics; 2010. https://www.ohe.org/publications/biosimilars-how-much-entry-and-price-competition-will-result. Accessed 8 Jan 2016.
Grabowski HG, Guha R, Salgado M. Biosimilar competition: lessons from Europe. Nat Rev Drug Discov. 2014;13(2):99–100. doi:10.1038/nrd4210.
van de Vooren K, Curto A, Garattini L. Biosimilar versus generic drugs: same but different? Appl Health Econ Health Policy. 2015;13:125–7.
IMS Institute for Healthcare Informatics. Assessing biosimilar uptake and competition in European markets. Parsippany: IMS Institute for Healthcare Informatics; 2014. https://www.imshealth.com/files/web/IMSH%20Institute/Healthcare%20Briefs/Assessing_biosimilar_uptake_and_competition_in_European_markets.pdf. Accessed 8 Jan 2016.
Bocquet F, Paubel P, Fusier I, Cordonnier A-L, Le Pen C, Sinegre M. Biosimilar granulocyte colony-stimulating factor uptakes in the EU-5 markets: a descriptive analysis. Appl Health Econ Health Policy. 2014;12:315–26.
Bocquet F, Paubel P, Fusier I, Cordonnier A-L, Sinegre M, Le Pen C. Biosimilar versus patented erythropoietins: learning from 5 years of European and Japanese experience. Appl Health Econ Health Policy. 2015;13:47–59.
Rovira J, Espin J, Garcia L, Olry de Labry A. The impact of biosimilars’ entry in the EU market. Report prepared for the European Commission (Directorate-General for Enterprise and Industry). Brussels: European Commission, 2011. http://ec.europa.eu/DocsRoom/documents/7651/attachments/1/translations/en/renditions/pdf. Accessed 23 July 2014.
Liefner M. Biosimilars: price dynamics in Europe. In: Mattison N, Mestre-Ferrandiz J, Towse A, editors. Biosimilars: how much entry and price competition will result? London: Office of Health Economics, 2010:51–9. https://www.ohe.org/publications/biosimilars-how-much-entry-and-price-competition-will-result. Accessed 8 Jan 2016.
Dylst P, Simoens S, Vulto A. Reference pricing systems in Europe: characteristics and consequences. GaBI J. 2012;1(3–4):127–31.
Stargardt T, Schreyögg J, Busse R. Pharmaceutical reference pricing in Germany: definition of therapeutic groups, price setting through regression procedure and effect [in German]. Gesundheitswesen. 2005;67(7):468–77.
Herr A, Moritz S. Pharmaceutical prices under regulation: co-payment exemptions and reference prices in Germany [DICE discussion paper no. 48]. Düsseldorf: Heinrich‐Heine‐Universität Düsseldorf; 2012. http://econstor.eu/bitstream/10419/57630/1/690258666.pdf. Accessed 7 Jan 2015.
GfK Market Access. Factors supporting a sustainable European biosimilar medicines market. Melton Mowbray: GfK NOP Limited; 2014. http://www.egagenerics.com/images/Website/GfK_Final_Report-_Factors_Supporting_a_Sustainable_European_Biosimilar_Medicines_Market.pdf. Accessed 8 Jan 2016.
Grabowski H. Biosimilar competition: lessons from Europe and prospects for the US [seminar briefing no. 15]. London: Office of Health Economics; 2014. https://www.ohe.org/publications/biosimilar-competition-lessons-europe-and-prospects-us. Accessed 7 Jan 2015.
Grabowski HG, Guha R, Salgado M. Regulatory and cost barriers are likely to limit biosimilar development and expected savings in the near future. Health Aff (Millwood). 2014;33(6):1048–57. doi:10.1377/hlthaff.2013.0862.
Curto A, Ghislandi S, van de Vooren K, Duranti S, Garattini L. Regional tenders on biosimilars in Italy: an empirical analysis of awarded prices. Health Policy. 2014;116(2–3):182–7.
Ortega E. Medicamentos biológicos de marca y biosimilares competirán en los mismos concursos públicos. Redacción Medica, 25 November 2014. http://www.redaccionmedica.com/secciones/industria/biologicos-de-marca-y-biosimilares-competiran-en-los-mismos-concursos-publicos-5934. Accessed 21 April 2015.
GaBI Online. Huge discount on biosimilar infliximab in Norway. Mol: Pro Pharma Communications International; 2015. http://www.gabionline.net/Biosimilars/General/Huge-discount-on-biosimilar-infliximab-in-Norway. Accessed 28 April 2015.
Benassi F. Norway’s discount on infliximab: a litmus test for biosimilar expansion in Europe? Englewood: IHS Life Sciences Blog; 2014. http://blog.ihs.com/norways-discount-on-infliximab-a-litmus-test-for-biosimilar-expansion-in-europe. Accessed 23 July 2014.
Diakonhjemmet Hospital. The NOR-SWITCH study [ClinicalTrials.gov study ID: NCT02148640]. Rockville Pike: National Library of Medicine; 2015. https://clinicaltrials.gov/ct2/show/study/NCT02148640. Accessed 8 Jan 2015.
Sint Maartenskliniek Nijmegen, Maartenskliniek Woerden, Radboud University Medical Center Nijmegen, Rijnstate Ziekenhuis. The effect of switching treatment from innovator infliximab to infliximab biosimilar on efficacy, safety and immunogenicity in patients with rheumatoid arthritis, spondyloarthritis or psoriatic arthritis in daily clinical care [Nederlands Trial Register no. NTR5279]. Amsterdam: Dutch Cochrane Centre; 2015. http://www.trialregister.nl/trialreg/admin/rctview.asp?TC=5279. Accessed 6 Oct 2015.
GaBI Online. Another infliximab switching trial started. Mol: Pro Pharma Communications International; 2015. http://gabionline.net/Biosimilars/News/Another‐infliximabswitching‐trialstarted?utm_source=GONL3&utm_campaign=d5217142de‐GONL+V15J02‐3&utm_medium=email&utm_term=0_735577ae74‐d5217142de‐77055973. Accessed 6 Oct 2015.
IMS Health Incorporated. Shaping the biosimilars opportunity: a global perspective on the evolving biosimilars landscape. London: IMS Health Incorporated; 2011. http://weinberggroup.com/pdfs/Shaping_the_biosimiliars_opportunity_A_global_perspective_on_the_evolving_biosimiliars_landscape.pdf. Accessed 8 Jan 2016.
Blackstone E, Fuhr J. Biopharmaceuticals: the economic equation. Biotechnol Health. 2007;4(6):41–5.
Kozlowski S, Woodcock J, Midthun K, Sherman RB. Developing the nation’s biosimilars program. N Engl J Med. 2011;365:385–8. doi:10.1056/NEJMp1107285.
Grabowski H, Kyle M. Generic competition and market exclusivity periods in pharmaceuticals. Manag Decis Econ. 2007;28(4–5):491–502.
Reiffen D, Ward M. Generic drug industry dynamics. Rev Econ Stat. 2005;87(1):37–49.
Saha A, Grabowski H, Birnbaum H, Greenberg P, Bizan O. Generic competition in the US pharmaceutical industry. Int J Econ Bus. 2006;13(1):15–38.
Towse A, Mestre-Ferrandiz J. The market for biosimilars: evolution and policy options. In: Mattison N, Mestre-Ferrandiz J, Towse A, editors. Biosimilars: how much entry and price competition will result? London: Office of Health Economics; 2010:25–35. https://www.ohe.org/publications/biosimilars-how-much-entry-and-price-competition-will-result. Accessed 8 Jan 2016.
Costa-Font J, McGuire A, Varol N. Price regulation and relative delays in generic drug adoption. J Health Econ. 2014;38:109.
Puig-Junoy J. Impact of European pharmaceutical price regulation on generic price competition: a review. Pharmacoeconomics. 2010;28(8):649–63.
Simoens S, De Coster S. Sustaining generic medicines markets in Europe. Leuven: Katholieke Universiteit Leuven Research Centre for Pharmaceutical Care and Pharmacoeconomics; 2006. http://www.assogenerici.org/articolihome/simoens-report_2006-04.pdf. Accessed 8 Jan 2016.
Kanavos P, Costa-Font J, Seeley E. Competition in off-patent drug markets: issues, regulation and evidence. Econ Policy. 2008;23(55):500–44. doi:10.1111/j.1468-0327.2008.00207.x.
BioMedTracker. Biosimilars special report. San Diego: Sagient Research Systems; 2015. http://www.biomedtracker.com/FreeReportDetail.cfm?ReportID=634&utm_source=email&utm_medium=email&utm_campaign=Biosimilars_free_report. Accessed 10 Oct 2015.
Marsh D, Kracov D, Kailasanath V, Wu W. Amgen V. Sandoz: Federal Circuit rules that the BPCIA’s “patent dance” is optional. Washington DC: Arnold and Porter LLP; 2015. http://www.arnoldporter.com/publications.cfm?action=advisory&u=iAmgenvSandoziFederalCircuitRulesThattheBPCIAsPatentDanceisOptional&id=1297. Accessed 11 Sept 2015.
Ahlstrom A, King R, Brown R, Clavdemans J, Mendelson D. Modeling federal cost savings from follow-on biologics. Washington DC: Avalere Health LLC; 2007.
Miller S, Houts J. Potential savings of biogenerics in the United States. St Louis: Express Scripts; 2007. https://www.pharmamedtechbi.com/~/media/Images/Publications/Archive/The%20Pink%20Sheet/69/008/00690080001/070219_express_scripts_generic_biologic_study.pdf. Accessed 8 Jan 2016.
Congressional Budget Office [CBO]. Cost estimate: S. 1695. Biologics Price Competition and Innovation Act of 2007. Washington, DC: CBO; 2008. https://www.cbo.gov/sites/default/files/cbofiles/ftpdocs/94xx/doc9496/s1695.pdf. Accessed 8 Jan 2016.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The Office of Health Economics receives a programme research grant from the Association of the British Pharmaceutical Industry. No separate funding was received for this paper.
Jorge Mestre-Ferrandiz, Adrian Towse and Mikel Berdud are all employees of the Office of Health Economics. Jorge Mestre-Ferrandiz and Adrian Towse, as part of the Office of Health Economics, received a research grant from Amgen to run a workshop on biosimilars and health technology assessment, and have previously received consulting funding from both innovator and biosimilar companies.
Author contributions
The article was conceived by Adrian Towse and Jorge Mestre-Ferrandiz. The literature review was conducted by Mikel Berdud. All authors contributed drafts for various parts of the paper, critically reviewing various iterations and approving the final draft submitted for publication.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Mestre-Ferrandiz, J., Towse, A. & Berdud, M. Biosimilars: How Can Payers Get Long-Term Savings?. PharmacoEconomics 34, 609–616 (2016). https://doi.org/10.1007/s40273-015-0380-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40273-015-0380-x