
Abstract
Edoxaban, a once daily non-vitamin K antagonist oral anticoagulant, is a direct, selective, reversible inhibitor of factor Xa (FXa). In healthy subjects, single oral doses of edoxaban result in peak plasma concentrations within 1.0–2.0 h of administration, followed by a biphasic decline. Exposure is approximately dose proportional for once daily doses of 15–150 mg. Edoxaban is predominantly absorbed from the upper gastrointestinal tract, and oral bioavailability is approximately 62 %. Food does not affect total exposure to edoxaban. The terminal elimination half-life in healthy subjects ranges from 10 to 14 h, with minimal accumulation upon repeat once daily dosing up to doses of 120 mg. The steady-state volume of distribution is approximately 107 L, and total clearance is approximately 22 L/h; renal clearance accounts for approximately 50 % of total clearance, while metabolism and biliary secretion account for the remaining 50 %. Intrinsic factors, such as age, sex and race, do not affect edoxaban pharmacokinetics after renal function is taken into account. Oral administration of edoxaban results in rapid changes in anticoagulatory biomarkers, with peak effects on anticoagulation markers (such as anti-FXa), the prothrombin time and the activated partial thromboplastin time occurring within 1–2 h of dosing.
Similar content being viewed by others
Edoxaban—a highly selective, competitive, concentration-dependent inhibitor of factor Xa—rapidly achieves peak plasma concentrations and exerts its peak effect on anticoagulatory biomarkers. |
The drug has high oral bioavailability, and its clearance involves both renal and non-renal pathways to almost equal extents. |
Coadministration with food does not affect total edoxaban exposure. |
Renal function affects exposure to edoxaban, but—independently—age, sex and race do not. |
1 Introduction
Anticoagulant therapy is commonly used for treatment and prevention of venous thromboembolism (VTE) [1] and prevention of stroke in patients with atrial fibrillation (AF) [2, 3]. The most common standard-of-care treatments include parenteral heparins and the oral vitamin K antagonist warfarin [1, 2].
The anticoagulant effect of warfarin relies on inhibition of vitamin K-dependent biosynthesis of four coagulation factors (prothrombin; and factors VII, IX and X) in the liver [4]. Warfarin has been used for treatment of VTE and stroke prevention in AF for more than 60 years, but its therapeutic utility has several limitations. These include a delayed onset of action, a narrow therapeutic index requiring close clinical monitoring of anticoagulation, an unpredictable and variable pharmacological response, and numerous drug–drug and food–drug interactions [4, 5]. Non-vitamin K antagonist oral anticoagulants (NOACs) were developed to inhibit single factors in the coagulation cascade and with pharmacological properties that overcome many of the limitations associated with warfarin use. These NOACs include apixaban, dabigatran, edoxaban and rivaroxaban.
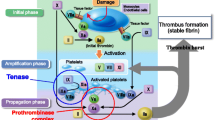
Edoxaban is an oral, selective, direct and reversible inhibitor of activated clotting factor X (FXa), the serine protease responsible for the generation of thrombin (Fig. 1) [6, 7]. Inhibition of FXa does not interfere with thrombin activity, which is involved in both coagulation and platelet activation. The efficacy and safety of edoxaban have been evaluated in two large phase 3 clinical trials for prevention of stroke and systemic embolic events in patients with non-valvular AF [8] and for treatment of deep vein thrombosis and pulmonary embolism [9]. Edoxaban was found to have efficacy noninferior to that of warfarin in both trials, with improved safety, assessed by bleeding events. Edoxaban 60 mg once daily has been approved for use for the above-mentioned indications in Japan [10], the USA [11], Switzerland [12] and the European Union [13]. In addition, edoxaban 30 mg is approved in Japan for prevention of VTE events in patients undergoing total knee replacement, total hip replacement or hip fracture surgery [10], on the basis of the results of three phase 3 trials in respective patient populations [14–16]. This manuscript reviews the pharmacokinetic and pharmacodynamic properties of edoxaban.

Site of edoxaban modulation in the coagulation cascade [3]
2 Physicochemical Properties
Edoxaban is the anhydrous free-drug form, with a molecular mass of 548.06. It is dosed as a monohydrate tosylate salt, which has a molecular weight of 738.27 [11, 17]. Edoxaban has low solubility in the pH range of 3–7, with a decrease in solubility with increasing pH [11]. In addition, its permeability in Caco-2 cells is poor [18], making it a Biopharmaceutics Classification System class IV compound [19].
3 Pharmacokinetics in Healthy Volunteers
The pharmacokinetics of edoxaban have been studied in many healthy volunteer studies. Single oral doses of 10–180 mg were administered to healthy subjects. Edoxaban exhibits generally linear and dose-proportional pharmacokinetics [20]. Following oral administration, edoxaban reaches peak plasma concentration (C max) values within 1–2 h. Edoxaban is widely distributed in the body, with a steady-state volume of distribution (V ss) (arithmetic mean ± standard deviation) of 107 ± 19.9 L [7]. The total clearance of edoxaban is estimated to be 21.8 ± 3.03 L/h, with renal and non-renal clearance contributing almost equally [7]. Renal clearance is estimated to be about 10.7 ± 3.00 L/h [7]. The terminal elimination half-life is approximately 10–14 h [11]. Shorter half-lives have been reported in some studies [20, 21]; however, because of a limited sampling duration, data from these studies should be interpreted with caution. A summary of the pharmacokinetic parameters of edoxaban is presented in Table 1.
Multiple-dose administration of once daily doses up to 120 mg results in minimal accumulation (accumulation ratio of about 1.1) [20], and the pharmacokinetics are linear and dose proportional—similar to the single-dose pharmacokinetics (Fig. 2). Steady state is achieved after 3 days of dosing. Both single- and multiple-dose administration result in C max within 1–2 h after dosing, followed by a biphasic decline (Fig. 3) [22].

Mean edoxaban plasma concentration–time profiles after single-dose administration: a linear scale and b logarithmic scale (data on file). The error bars represent the standard deviation

Mean edoxaban concentration–time profiles after administration of edoxaban 60 mg once daily after a single dose (day 1; N = 23) or at steady state (day 4; N = 23): a linear scale and b logarithmic scale [22]. The error bars represent the standard deviation
3.1 Absorption
Edoxaban is the active moiety and the predominant circulating drug-related moiety. Oral bioavailability is 62 % [7]. Edoxaban is primarily absorbed in the upper gastrointestinal tract, with approximately 13 % absorbed in the colon [23]. In a single-dose, food–effect study in healthy volunteers, the mean peak exposure (C max) from a 60 mg dose increased in the fed state by 6–22 %, in comparison with the fasted state, with a high degree of individual variability [24]. This variability likely results from the effect of food on the rate of absorption; the median t max (minimum–maximum) increased from 1 h (0.50–2.50 h) in the fasted state to 1.50 h (0.50–5.0 h) in the fed state. The effect of food on the total exposure was minimal and less variable, increasing by 12–15 %, and was not considered clinically significant [24]. In a healthy subject study, administration of edoxaban 2 h after esomeprazole at steady state had no significant effect on the total exposure, with the 90 % confidence interval (CI) of the ratio of the least-squares mean being between 80 and 125 %, although the peak exposure decreased by about 33 % [11].
3.2 Distribution
The disposition of edoxaban is biphasic and is described by a 2-compartment model [11, 25]. The (mean ± standard deviation) V ss is 107 ± 19.9 L [11].
3.3 Metabolism, Transport, Protein Binding and Distribution to Blood
In vitro, five phase 1 edoxaban metabolites were detected in human liver microsomes: M-1, M-4, M-5, M-6 and a hydroxylated metabolite at the N,N-dimethylcarbamoyl group of edoxaban (hydroxymethyl edoxaban) (M-7) [26]. The formation of the human-unique metabolite M-4 is catalysed by carboxylesterase-1 (CES1) present in human liver microsomes and in the cytosol [27]. Cytochrome P450 isoenzyme (CYP) 3A4 mediates the formation of M-5 and hydroxymethyl edoxaban in the presence of nicotinamide adenine dinucleotide phosphate (NADPH) [26]. M-8, a minor metabolite, is postulated to arise spontaneously (non-enzymatically) through an intermediary, hydroxymethyl edoxaban, formed via CYP3A4/5 [21].
In healthy human subjects, six phase 1 metabolites (M-1, M-2, M-4, M-5, M-6 and M-8) and a glucuronide (M-3) were detected in plasma (Fig. 4). The human-unique metabolite M-4 is the major metabolite but is present at less than 10 % of the total edoxaban exposure in healthy adults [21]. A summary of the pharmacokinetic parameters is presented in Table 1. Phase 2 metabolism is mediated by glucuronidation to form an N-glucuronide metabolite (M-3). This metabolite has not been quantified. Three of the metabolites (M-4, M-6 and M-8) have anticoagulant activity, with half-maximal inhibitory concentration (IC50) values for anti-FXa of 1.8 nM (M-4), 6.9 nM (M-6) and 2.7 nM (M-8). The IC50 of edoxaban for anti-FXa is 3 nM [17]. However, because of its low abundance and high protein binding (80 %), the most abundant metabolite, M-4, is not expected to contribute significantly to the overall pharmacological activity of edoxaban in subjects with at least moderate renal function [28]. Other metabolites are present in even lower abundance and, in the absence of CYP inducers, do not contribute significantly to overall anticoagulant activity (Table 2). None of the metabolic pathways individually contribute greater than 10 % to total clearance [21].

Postulated edoxaban metabolism [21]. CES1 carboxylesterase-1, CYP3A4/5 cytochrome P450 isoenzyme 3A4/5, HF human faeces, HP human plasma, HU human urine, M metabolite, UGT urine 5′-diphospho-glucuronosyltransferase
In vitro bidirectional transport assays in human colon adenocarcinoma Caco-2 cell monolayers reveal that edoxaban is a substrate of the efflux transporter P-glycoprotein (P-gp) but is not a substrate for uptake transporters, such as organic anion-transporting polypeptide (OATP) 1B1, organic anion transporters (OATs) 1 and 3, or organic cation transporter (OCT) 2 [18]. Exploratory pharmacogenomic analyses demonstrate no effect of the ATP-binding cassette, subfamily B, member 1 genotype (C3435T) on the pharmacokinetics of edoxaban [29].
Edoxaban has a minimal inhibitory effect on CYPs (IC50 ≥100 μM). It shows a weak inhibitory effect on P-gp, OATP1B1 and OATP1B3 (IC50 ≥50 μM), and a minimal effect on other drug transporters (OAT1, OAT3, OCT1 and OCT2; IC50≥100 μM) [18]. In human hepatocytes, edoxaban and the metabolites M-4 and M-1 do not induce gene expression of CYP1A2, CYP3A4 or multidrug resistance protein 1 (MDR1; P-gp) (data on file). Thus, at clinical concentrations, edoxaban is unlikely to affect the pharmacokinetics of other drugs that are substrates of CYPs or transporters, such as P-gp, OATP1B1, OATP1B3, OAT1, OAT3, OCT1 or OCT2.
The in vitro total plasma protein binding of edoxaban at concentrations of 0.2–5 µg/mL is about 55 %, whereas the human-unique metabolite M-4 is approximately 80 % bound to plasma proteins over a concentration range of 0.2–2 µg/mL [11, 17]. Edoxaban partitions almost equally in blood (46 %) and plasma [17].
3.4 Elimination
Edoxaban is primarily eliminated unchanged in urine and through biliary secretion, with metabolism contributing to a lesser extent towards total clearance [21] (Fig. 5). Renal clearance of unchanged drug contributes approximately 50 % to total clearance [7], with the remaining 50 % non-renal clearance occurring through metabolism and biliary secretion. In healthy subjects administered 60 mg of [14C]-edoxaban, 62 and 35 % of the total radioactive dose was detected in faeces and urine, respectively. Specifically, unchanged edoxaban accounted for 49 and 24 % of the total administered radioactive dose detected in faeces and urine, respectively [21]. The human-unique metabolite M-4 was detected in small amounts in both urine and faeces, but was not quantified [21]. The amounts of M-1, M-6 and M-8 in faeces and urine ranged from below the limit of quantification to 1.66 and to 1.85 % of total radioactivity, respectively [21].

Schematic of clearance pathways for edoxaban in adults. CES-1 carboxylesterase-1, CYP3A4/5 cytochrome P450 isoenzyme 3A4/5
Data from bile cannulation studies in rats also suggest biliary excretion and enterohepatic recirculation (data on file). Furthermore, mass balance data and pharmacokinetic data from the oral versus intravenous administration study [7, 21] indicate biliary excretion of edoxaban. The oral half-life of edoxaban (10–14 h) [11] suggests distribution and redistribution resulting in a higher terminal-phase volume of distribution, possibly due to enterohepatic recirculation.
3.5 Dose and Time Dependency
A pooled analysis of phase 1 data from five studies—with doses of 10, 15, 30, 60, 90, 120, 150 and 180 mg—indicates a slightly less-than-dose-proportional dose–exposure relationship [25]. The population estimate of model-based relative bioavailability (F 1) was found to remain constant for the dose range of 10–30 mg but decreased with increasing doses at higher dose levels. For doses above 30 mg, every 30 mg increase in dose was associated with a 6.7 % decrease in F 1, presumably due to a decreased dissolution rate [25]. This is in line with the absolute bioavailability of 62 % for a 60 mg oral dose as determined by Matsushima et al. [7] and thus is not considered clinically significant.
3.6 Variability
Inter- and intra-subject variability in the clearance and volume of distribution of edoxaban is low (<30 %) [17].
4 Effects of Intrinsic Factors on Edoxaban Pharmacokinetics
The effects of various intrinsic factors on the pharmacokinetics of edoxaban have been evaluated in many studies [30–32] and in population pharmacokinetic analyses [11, 33] of patient data. In general, renal function is the most important intrinsic determinant of total exposure. After accounting for renal function, age is no longer an independent factor affecting exposure to edoxaban. Increasing body weight is associated with decreasing exposure. After accounting for body weight, sex is no longer an independent determinant of the pharmacokinetics of edoxaban. Similarly, after accounting for body weight, edoxaban exposure is similar in Asians and non-Asians [11, 17]. The data on other racial subgroups are limited.
4.1 Renal Function
Renal clearance is a major component of total clearance of edoxaban; hence, the pharmacokinetics of edoxaban have been evaluated in subjects with varying degrees of renal impairment and normal renal function. The total exposure to edoxaban increases with increasing degrees of renal impairment, with 32, 74 and 72 % higher exposure in subjects with mild [creatinine clearance (CLCR) ≥50–≤80 mL/min], moderate (CLCR ≥30–<50 mL/min) and severe (CLCR <30 mL/min) renal impairment, respectively, versus subjects with normal renal function [31]. Total exposure and peak exposure data are provided in Table 3. In subjects with end-stage renal disease undergoing peritoneal dialysis, the total exposure is 93 % higher [31]. Decreasing renal clearance results in higher exposure, but as renal clearance is only one component of total clearance, there is a plateauing of the impact of renal impairment on total clearance. Subjects with moderate and severe renal impairment have generally similar total clearance values, despite lower renal clearance (Table 3). In these subjects, metabolic clearance plays the predominant role in total clearance of edoxaban.
In subjects with renal impairment, the total exposure to the metabolites (M-1, M-4, and M-6) increases with increasing severity of renal impairment. In comparison with subjects with normal renal function, the total exposure to edoxaban metabolites in mild, moderate and severe renal impairment is higher: exposure to M-1 is 1.52-, 2.94- and 5.39-fold higher, respectively; exposure to M-4 is 2.25-, 3.74- and 3.91-fold higher, respectively; and exposure to M-6 is 1.65-, 2.85- and 2.52-fold higher, respectively (data on file). The relative abundance of metabolites with respect to edoxaban increases with the degree of renal impairment.
4.2 Hepatic Function
Impaired hepatic function could alter hepatic metabolism and affect exposure to edoxaban, as well as blood coagulation. The impact of hepatic function on the single-dose pharmacokinetics was assessed in subjects with mild and moderate hepatic impairment and matched healthy controls [32]. Hepatic impairment did not significantly impact the peak or total exposure in subjects with mild or moderate hepatic impairment, consistent with a limited role of hepatic metabolism in total clearance of edoxaban.
5 Effects of Extrinsic Factors on Edoxaban Pharmacokinetics
As previously described, edoxaban is metabolized by CES1 (<10 %), by CYP3A4 (<10 %) and via glucuronidation; however, metabolism is a minor clearance pathway for edoxaban in subjects with normal renal function. Hence, inhibitors or inducers of these enzymes are unlikely to have a clinically relevant interaction with edoxaban.
Drug interaction studies were nonetheless performed to investigate the effects of CYP3A4 inhibitors on the pharmacokinetics of edoxaban. Additionally, the effects of other drugs that could be dosed concomitantly with edoxaban were evaluated. Since edoxaban is a substrate of the efflux transporter P-gp, several drug interaction studies were conducted with P-gp inhibitors, substrates and inducers. A summary of the drug interaction studies exploring the effects of CYP and transporter inhibition/induction on the pharmacokinetics of edoxaban is presented in Table 4. The effect of concomitant P-gp inhibitor use was an increase in exposure to edoxaban [the maximum observed plasma drug concentration (C max) and the area under the concentration–time curve (AUC)], but the increase was less than twofold. Drugs that are inhibitors of P-gp and strong inhibitors of CYP3A4/5 (e.g. ketoconazole, erythromycin) did not result in a greater increase in exposure than drugs that are inhibitors of P-gp and moderate (e.g. verapamil) or mild inhibitors (e.g. cyclosporine) of CYP3A4/5, thereby reinforcing the fact that metabolism by CYP3A4/5 is not a major clearance pathway for edoxaban. These results are summarized below and in Fig. 6.

Summary of edoxaban drug interaction studies results [11]. The triangles represent the geometric least-squares mean ratio of the maximum observed plasma drug concentration (C max) when edoxaban was dosed alone or with the test drug, the squares represent the geometric least-squares mean ratio for the area under the concentration–time curve (AUC) and the horizontal bars represent the 90 % confidence interval. P-gp P-glycoprotein
-
Ketoconazole (inhibitor of P-gp transporter; strong CYP3A4 inhibitor): coadministration of ketoconazole increased the single-dose peak and total exposure to edoxaban by 89 and 87 %, respectively [34].
-
Quinidine (inhibitor of P-gp and OCT2 transporters; strong inhibitor of CYP2D6): coadministration of oral quinidine increased the single-dose peak and 24-h exposure to orally dosed edoxaban by 85 and 77 %, respectively [37].
-
Orally administered quinidine increased the peak and total exposure to intravenous edoxaban by 7.5 and 35 %, respectively [7]. Since edoxaban will not be available as an intravenous formulation, this study provides mechanistic insight but is not meant for dosing guidance.
-
Verapamil [inhibitor of P-gp (main effect); moderate inhibitor of CYP3A4]: coadministration of sustained-release verapamil increased the peak and 24-h exposure to single doses of edoxaban by 53 % [37].
-
Erythromycin (inhibitor of P-gp; moderate CYP3A4 inhibitor): coadministration of erythromycin increased the peak and total exposure to single doses of edoxaban by 68 and 85 %, respectively [38].
-
Cyclosporine [inhibitor of P-gp, OATP1B1 and breast cancer resistance protein (BCRP); weak CYP3A4 inhibitor]: coadministration of cyclosporine increased both the peak and total exposure to single doses of edoxaban by 74 and 73 %, respectively [39].
-
Dronedarone (inhibitor of P-gp): coadministration of dronedarone increased the peak and total exposure to single doses of edoxaban by 46 and 85 %, respectively [37].
-
Amiodarone (inhibitor of P-gp; moderate CYP2C9 inhibitor, weak CYP2D6 inhibitor): coadministration of amiodarone, following 3 days of once daily dosing, increased the peak and total exposure to single doses of edoxaban by 66 and 40 %, respectively [37]. Amiodarone has a long half-life of 58 days (range 15–142 days) [40]; the study design was not adequate to achieve steady-state (or maximal) exposure to amiodarone [41].
-
Rifampin, an inducer of P-gp (strong inducer of CYP3A4; moderate inducer of CYP2B6, 2C8, 2C9, 2C19; inhibitor of P-gp, OATP1B1, OATP1B3) after 7 days of dosing, decreased total edoxaban exposure by about 34 %, with no effect on the peak exposure [42].
Other drug interaction study results with potential concomitant medications are summarized below and in Fig. 6.
-
Digoxin (P-gp substrate): coadministration of digoxin increased the edoxaban C max by 16 %, with no significant effect on total exposure or renal clearance at steady state [37].
-
Atorvastatin (substrate of OATP1B1 and OATP1B3; weak inhibitor of CYP3A4): coadministration of atorvastatin with edoxaban had no effect on the peak or total exposure to edoxaban [37].
-
Naproxen: coadministration of naproxen and edoxaban had no effect on the peak and total exposure to edoxaban [43]. Coadministration of naproxen and edoxaban resulted in increased prolongation of bleeding time in comparison with either drug dosed alone. Coadministration of naproxen increased the baseline-corrected ratio of bleeding time by 72 % on day 2 in comparison with edoxaban alone (with a 90 % CI of 139.3–213.3). In contrast, coadministration of edoxaban with naproxen increased the equivalent bleeding time by 22 % in comparison with naproxen alone (with a 90 % CI of 98.1–151.0) [43]. Naproxen decreased the baseline-corrected ratio for platelet aggregation [day 2 ratio (90 % CI): 69.89 % (68.20–71.62)], while edoxaban did not affect the platelet aggregation effects of naproxen—the baseline-corrected ratios were comparable for the two treatments [day 2 ratio (90 % CI): 97.20 (94.81–99.64)] (data on file).
-
Aspirin: coadministration of high-dose aspirin (325 mg) increased the steady-state peak and total exposure to edoxaban by 34 and 30 % [43], respectively, and reduced renal clearance by 17 % (data on file), possibly because of inhibition of active renal secretion. Coadministration of low-dose aspirin (100 mg) did not affect the peak or total exposure to edoxaban either after single-dose administration or at steady state, as the 90 % CIs were entirely contained within the 80–125 % bounds [43]. Coadministration of edoxaban and low-dose (100 mg) or high-dose aspirin (325 mg) resulted in an additive effect on the prolongation of bleeding time [43]. The anticoagulant effects of edoxaban were not affected by coadministration of aspirin. Coadministration of low-dose aspirin (100 mg) did not significantly affect the prothrombin time (PT), international normalized ratio (INR), activated partial thromboplastin time (aPTT) or intrinsic FX activity [43].
-
Enoxaparin: enoxaparin did not affect the peak and total exposure to edoxaban when dosed concomitantly or 12 h apart. Coadministration of edoxaban 60 mg and subcutaneous enoxaparin 1 mg/kg resulted in an increased effect on thrombin generation assay parameters in comparison with either drug dosed alone [44]. The effect, in general, was not additive, with the exception of the thrombin generation lag time and time to peak. The effect on anti-FXa, when both drugs were coadministered, appeared to be additive [44].
-
Esomeprazole, a proton pump inhibitor, which increases gastric pH to approximately 5 after multiple doses, increased the C max of edoxaban dosed with a 2-h stagger at steady state by approximately 33 % (90 % CI 57.98–78.07), while the total exposure was within the 80–125 % CI [11], demonstrating no significant effect on the total exposure (data on file). A previous study demonstrated no significant effect on the peak and total exposure with concurrent dosing [45]. Of note, the dissolution and solubility of edoxaban are pH dependent; hence, in patients with achlorhydria or other conditions (or concomitant use of drugs) that increase stomach pH above 6, the rates of solubility and dissolution of edoxaban may be reduced, thereby affecting total absorption of edoxaban.
6 Pharmacology and Pharmacodynamics
Serine protease FXa binds coagulation factor Va on the surface of activated platelets and forms a prothrombinase complex, which subsequently converts prothrombin to thrombin [46]. FXa is the primary site of prothrombin amplification and thus constitutes an attractive target for anticoagulant treatment (Fig. 1). Edoxaban is a selective and competitive inhibitor of human FXa, with an inhibitory constant (Ki) value of 0.561 nM [6]. It inhibits the generation of thrombin and formation of blood clots in a dose-dependent manner.
Edoxaban produces rapid anticoagulation with a direct concentration–effect relationship. Within 1–2 h after single-dose administration to healthy subjects, maximum dose-proportional effects of edoxaban 10–150 mg on plasma anti-FXa activity were observed, concurrent with the edoxaban C max [20]. Plasma edoxaban concentrations are linearly related to anti-FXa activity in healthy subjects (Fig. 7). A similar linear relationship was observed in patients with VTE and AF.

Observed anti-factor Xa (anti-Fxa) activity versus plasma edoxaban concentration after administration of a single 60 or 180 mg dose in healthy subjects (data on file)
Intrinsic FX activity data from a phase 2 study of patients with AF [47] were modelled. Using a logistic regression model and breakpoint or threshold concepts, it was demonstrated that the duration of intrinsic FX suppression exceeding a threshold—as opposed to the actual magnitude of suppression—correlated with bleeding risk [47]. The model identified the most significant predictor for bleeding as the period of time during which intrinsic FX activity was maintained at 15 % or less. Prolonged suppression below this threshold accounted for more bleeding observed in the phase 2 study, where, despite the same total daily dose from a 60 mg once daily regimen versus a 30 mg twice daily regimen, the 30 mg twice daily regimen resulted in more bleeding [48, 49]; the 30 mg twice daily regimen suppressed intrinsic FX activity for 18.8 h, while the 60 mg once daily regimen led to 13.7 h of intrinsic FX activity suppression [47].
In the Badimon Perfusion-Chamber model, a single 60 mg dose of edoxaban in healthy subjects markedly reduced ex vivo thrombus formation under high and low shear rates, consistent with anti-FXa activity [50]. Also in healthy subjects, a shed blood model showed significant inhibition of thrombin generation and platelet activation by edoxaban at single doses of 30, 60 and 120 mg [51]. A nadir for shed blood levels of prothrombin fragments 1 + 2 (F 1+2) and for platelet activation-marker β-thromboglobulin were achieved at 1.5 h postdose, whereas placebo had minimal suppressive effects. The anticoagulant effects of edoxaban were sustained for 24 h [51]. In a study of elderly healthy subjects, in addition to producing anti-FXa activity and reducing F1+2, edoxaban 60 mg twice daily induced a thrombin generation lag time and inhibited the peak and constant velocity indices. There was no evidence of a rebound effect following cessation of dosing [52].
Other coagulation markers affected by edoxaban include PT and aPTT. In healthy subjects treated with edoxaban 60 mg daily for 4 days, PT and aPTT reached peak elevations within 2 h and returned to near-baseline levels within 24 h, both after a single dose (day 1) and after multiple doses (day 4) [53]. However, modest magnitudes of change and variability make none of these tests ideal for routine and consistent clinical assessment of drug effects.
7 Cardiovascular Safety
The effect of edoxaban on cardiac polarization was evaluated in an active and placebo-controlled ‘thorough QTc’ [54] study of supratherapeutic doses of 90 and 180 mg in healthy subjects [55]. Edoxaban did not have a threshold pharmacological effect on cardiac repolarization. The upper bounds of the 95 % one-sided CIs for the least-squares mean of the placebo-corrected QTc interval (QTcI) change from time-matched baseline did not exceed 4 ms at any time point after dosing of either dose. The absolute value of QTcI did not exceed 450 ms for any subject on either dose of edoxaban, and all except one subject had changes in QTcI that were below 30 ms at all time points [17, 55]. One subject in the 90 mg edoxaban dose group had a change in QTcI value of 32 ms at 10 h postdose. No clinically relevant changes in electrocardiogram waveforms were noted with edoxaban treatment. A positive correlation between plasma concentrations and change in QTcI was not observed up to edoxaban concentrations of 857 ng/mL [55].
8 Switching
Edoxaban is the fourth entrant into the marketplace, and patients might switch from other anticoagulants to edoxaban. On the basis of the single-dose pharmacokinetics of edoxaban and pharmacodynamic effects, which generally parallel the plasma concentration–time profiles, it appears that switching from other anticoagulants to edoxaban is feasible at the next scheduled dosing time of the prior treatment. This maintains adequate anticoagulation. Thus, switching from warfarin to edoxaban 24 h after cessation of warfarin therapy maintains continued anticoagulation with no effect of warfarin on the pharmacokinetics of edoxaban [56]. Similarly, switching from enoxaparin to edoxaban could be performed at the next scheduled dosing time for enoxaparin [44]. For patients receiving unfractionated heparin, it is recommended that edoxaban is started 4 h after stopping the heparin infusion [11].
The effects of switching from the other NOACs to edoxaban on edoxaban pharmacokinetics and pharmacodynamics have also been studied. A single oral dose of edoxaban 60 mg administered on day 4, following administration of once daily rivaroxaban 20 mg for 3 days, resulted in pharmacokinetic parameters similar to those of continual treatment with edoxaban [22]. Anticoagulant effects after switching from rivaroxaban to edoxaban, as assessed by assays such as anti-FXa, PT, aPTT and endogenous thrombin potential (ETP), were also similar to those after 4 days of continual edoxaban treatment [53]. Similar results were obtained after switching to edoxaban 60 mg after 3 days of dosing with apixaban 5 mg twice daily [17]. Finally, edoxaban pharmacokinetic parameters were similar after switching from dabigatran 150 mg twice daily for 3 days to an edoxaban 60 mg single dose, compared with 4 days of continual treatment with edoxaban alone [22]. Upon analysis of anticoagulant effects, measurements of aPTT and ETP suggested a carryover effect of dabigatran on anticoagulation, although PT and anti-FXa activities were not different between subjects who switched treatment from dabigatran and those who received continuous treatment with edoxaban alone [53]. Thus, switching from rivaroxaban or dabigatran to edoxaban could also be performed at the next scheduled time of dosing.
Appropriate transitioning from edoxaban to warfarin can be likely achieved by maintaining optimal anticoagulation via two options: by coadministration of a parenteral anticoagulant and warfarin after cessation of edoxaban dosing until INR ≥2.0, at which time parenteral anticoagulation can be stopped, or by coadministration of a 50 % lower dose of edoxaban along with warfarin until INR ≥2.0, at which time the reduced dose of edoxaban can be stopped. With either option, INR should be checked weekly [11, 57]. To switch from edoxaban to another NOAC, administration of the NOAC should begin at the next scheduled dose of edoxaban [11]. Clinical evaluation of this scheme, as patients transitioned off blinded edoxaban or warfarin to the open-label anticoagulant, minimized the number of major bleeding and stroke events during the 30 days of posttrial follow-up [57].
9 Summary and Conclusion
Edoxaban—a highly selective, competitive, concentration-dependent inhibitor of FXa—exhibits rapid achievement of C max and peak effects on biomarkers such as PT, aPTT and anti-FXa. It has high oral bioavailability, and its clearance mechanisms involve both renal and non-renal pathways to almost equal extents. P-gp inhibitor drugs increase the total exposure to less than twofold, while CYP3A4 inhibition causes a minimal additional change in exposure. Administration of food with edoxaban does not affect total exposure. Renal function affects exposure to edoxaban, but—independently—age, sex and race do not. Edoxaban concentrations have been shown to have a linear correlation with anti-FXa activity. Although edoxaban has active metabolites, because of their low abundance, they are unlikely to contribute significantly to anticoagulant activity in most patients.
References
Kearon C, Akl EA, Comerota AJ, Prandoni P, Bounameaux H, Goldhaber SZ, et al. Antithrombotic therapy for VTE disease: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;141(2 Suppl):e419S–94S.
Camm AJ, Lip GY, De Caterina R, Savelieva I, Atar D, Hohnloser SH, et al. 2012 focused update of the ESC guidelines for the management of atrial fibrillation: an update of the 2010 ESC guidelines for the management of atrial fibrillation. Developed with the special contribution of the European Heart Rhythm Association. Eur Heart J. 2012;33(21):2719–47.
January CT, Wann LS, Alpert JS, Calkins H, Cigarroa JE, Cleveland JC Jr, et al. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. Circulation. 2014;130(23):2071–104.
Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther M, Palareti G, et al. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest. 2008;133(6 Suppl):160S–98S.
Wells PS, Holbrook AM, Crowther NR, Hirsh J. Interactions of warfarin with drugs and food. Ann Intern Med. 1994;121(9):676–83.
Furugohri T, Isobe K, Honda Y, Kamisato-Matsumoto C, Sugiyama N, Nagahara T, et al. DU-176b, a potent and orally active factor Xa inhibitor: in vitro and in vivo pharmacological profiles. J Thromb Haemost. 2008;6(9):1542–9.
Matsushima N, Lee F, Sato T, Weiss D, Mendell J. Bioavailability and safety of the factor Xa inhibitor edoxaban and the effects of quinidine in healthy subjects. Clin Pharm Drug Dev. 2013;2:358–66.
Giugliano RP, Ruff CT, Braunwald E, Murphy SA, Wiviott SD, Halperin JL, et al. Edoxaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2013;369(22):2093–104.
The Hokusai VTE Investigators, Buller HR, Decousus H, Grosso MA, Mercuri M, Middeldorp S, et al. Edoxaban versus warfarin for the treatment of symptomatic venous thromboembolism. N Engl J Med. 2013;369(15):1406–15.
Lixiana® (edoxaban tosilate hydrate tablets): full prescribing information. Tokyo: Daiichi Sankyo Company, Ltd.; 2014.
Savaysa™ (edoxaban) tablets for oral use: full prescribing information. Parsippany: Daiichi Sankyo Inc.; 2015.
Daiichi Sankyo’s once-daily Lixiana® (edoxaban) approved for the prevention of stroke and systemic embolism in non-valvular atrial fibrillation and for the treatment and prevention of recurrent venous thromboembolism in Switzerland [press release]. April 15, 2015. Available at www.daiichisankyo.com/media_investors/media_relations/press_releases/detail/006269.html.
Lixiana® (edoxaban): package leaflet. Munich: Daiichi Sankyo Europe GmbH; 2015.
Fuji T, Fujita S, Kawai Y, Nakamura M, Kimura T, Kiuchi Y, et al. Safety and efficacy of edoxaban in patients undergoing hip fracture surgery. Thromb Res. 2014;133(6):1016–22.
Fuji T, Wang CJ, Fujita S, Kawai Y, Kimura T, Tachibana S. Safety and efficacy of edoxaban, an oral factor Xa inhibitor, for thromboprophylaxis after total hip arthroplasty in Japan and Taiwan. J Arthroplasty. 2014;29(12):2439–46.
Fuji T, Fujita S, Kawai Y, Nakamura M, Kimura T, Fukuzawa M, et al. Efficacy and safety of edoxaban versus enoxaparin for the prevention of venous thromboembolism following total hip arthroplasty: STARS J-V. Thromb J. 2015;13:27.
Daiichi Sankyo, Inc. Savaysa (edoxaban tosylate): FDA Cardiovascular and Renal Drugs Advisory Committee briefing document. NDA 206316. Meeting date: October 30, 2014. Available at: http://www.fda.gov/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/CardiovascularandRenalDrugsAdvisoryCommittee/ucm420703.htm. Accessed 5 Nov 2015.
Mikkaichi T, Yoshigae Y, Masumoto H, Imaoka T, Rozehnal V, Fischer T, et al. Edoxaban transport via P-glycoprotein is a key factor for the drug’s disposition. Drug Metab Dispos. 2014;42(4):520–8.
Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12(3):413–20.
Ogata K, Mendell-Harary J, Tachibana M, Masumoto H, Oguma T, Kojima M, et al. Clinical safety, tolerability, pharmacokinetics, and pharmacodynamics of the novel factor Xa inhibitor edoxaban in healthy volunteers. J Clin Pharmacol. 2010;50(7):743–53.
Bathala MS, Masumoto H, Oguma T, He L, Lowrie C, Mendell J. Pharmacokinetics, biotransformation, and mass balance of edoxaban, a selective, direct factor Xa inhibitor, in humans. Drug Metab Dispos. 2012;40(12):2250–5.
Parasrampuria D, Weilert J, Maa J, He L, Shi M, Brown K. Pharmacokinetics (PK) of edoxaban, a novel oral anticoagulant (NOAC), when dosed alone or following switching from dabigatran or rivaroxaban. Clin Pharmacol Ther. 2014;95(Suppl 1):S83.
Parasrampuria DA, Kanamaru T, Connor A, Wilding I, Ogata K, Shimoto Y, et al. Evaluation of regional gastrointestinal absorption of edoxaban using the enterion capsule. J Clin Pharmacol. 2015 [Epub ahead of print].
Mendell J, Tachibana M, Shi M, Kunitada S. Effects of food on the pharmacokinetics of edoxaban, an oral direct factor Xa inhibitor, in healthy volunteers. J Clin Pharmacol. 2011;51(5):687–94.
Yin OQ, Miller R. Population pharmacokinetics and dose–exposure proportionality of edoxaban in healthy volunteers. Clin Drug Investig. 2014;34(10):743–52.
Masumoto H, Yoshigae Y, Watanabe K, Takakusa H, Okazaki O, Izumi T. In vitro metabolism of edoxaban and the enzymes involved in the oxidative metabolism of edoxaban. AAPS J. 2010;12 (Suppl 2):Abstract W4308.
Ogura Y, Masumoto H, Yoshigae Y, Kosaka T, Shinagawa A. Identification of human hydrolase involved in the metabolism of edoxaban to a human major metabolite. AAPS J. 2010;12:Abstract W4298.
Jonsson S, Simonsson US, Miller R, Karlsson MO. Population pharmacokinetics of edoxaban and its main metabolite in a dedicated renal impairment study. J Clin Pharmacol. 2015 May 12 [Epub ahead of print].
Vandell A, Lee J, Shi M, Rubets I, Brown K, Walker JR. Lack of an affect of the ABCB1 C3435T (RS104564) polymorphism on the pharmacokinetics of edoxaban, a novel factor Xa inhibitor. Clin Pharmacol Therap. 2014;95(Suppl 1):S74.
Mendell J, Shi M. Safety, tolerability, pharmacokinetic (PK) and pharmacodynamic (PD) profiles of edoxaban in healthy post-menopausal or surgically sterile females, and healthy elderly males. Eur Heart J. 2011;32(Suppl 1):461.
Ridout G, de la Motte S, Niemczyk S, Sramek P, Johnson L, Jin J, et al. effect of renal function on edoxaban pharmacokinetics and on population PK model. J Clin Pharmacol. 2009;49:1091–130 (Abstract 144).
Mendell J, Johnson L, Chen S. An open-label, phase 1 study to evaluate the effects of hepatic impairment on edoxaban pharmacokinetics and pharmacodynamics. J Clin Pharmacol. 2015 May 22 [Epub ahead of print].
Yin OQ, Tetsuya K, Miller R. Edoxaban population pharmacokinetics and exposure-response analysis in patients with non-valvular atrial fibrillation. Eur J Clin Pharmacol. 2014;70(11):1339–51.
Zahir H, Mendell J, Jin J, He L, Parasrampuria DA. The effect of ketoconazole on the pharmacokinetics and pharmacodynamics of edoxaban in healthy adults. Clin Pharm Drug Dev. 2014;(Suppl 1):1–59.
FDA Center for Drug Evaluation Research. FDA draft guidance for industry: drug interaction studies—study design, data analysis, implications for dosing, and labeling recommendations. 2012. Available at: http://www.fda.gov/downloads/Drugs/Guidances/ucm292362.pdf. Accessed 17 Apr 2015.
Flockhart DA. Drug interactions: cytochrome P450 drug interaction table. Indiana University School of Medicine. 2009. Available at: https://static.medicine.iupui.edu/divisions/clinpharm/content/p450_Table_Oct_11_2009.pdf. Accessed 20 Apr 2015.
Mendell J, Zahir H, Matsushima N, Noveck R, Lee F, Chen S, et al. Drug–drug interaction studies of cardiovascular drugs involving P-glycoprotein, an efflux transporter, on the pharmacokinetics of edoxaban, an oral factor Xa inhibitor. Am J Cardiovasc Drugs. 2013;13(5):331–42.
Mendell J, Zhang G, He L, Parasrampuria DA. The effects of erythromycin on the pharmacokinetics and pharmacodynamics of edoxaban. Clin Pharmacol Drug Dev. 2014;3(Suppl 1):1–59.
Matsushima N, Zahir H, Mendell J, Chen S, He L, Parasrampuria DA. Coadministration of cyclosporine increased edoxaban exposure in healthy adults. Clin Pharmacol Drug Dev. 2014;3(Suppl 1):1–59.
Cordarone® (amiodarone HCl) tablets: full prescribing information. Wyeth Pharmaceuticals Inc., a subsidiary of Pfizer Inc.; Philadelphia. 2014. Available at: http://labeling.pfizer.com/showlabeling.aspx?id=93. Accessed 28 Mar 2015.
Robinson K, Johnston A, Walker S, Mulrow JP, McKenna WJ, Holt DW. The digoxin–amiodarone interaction. Cardiovasc Drugs Ther. 1989;3(1):25–8.
Mendell J, Chen S, He L, Desai M, Parasramupria DA. The effect of rifampin on the pharmacokinetics of edoxaban in healthy adults. Clin Drug Investig. 2015;35(7):447–53.
Mendell J, Lee F, Chen S, Worland V, Shi M, Samama MM. The effects of the antiplatelet agents, aspirin and naproxen, on pharmacokinetics and pharmacodynamics of the anticoagulant edoxaban, a direct factor Xa inhibitor. J Cardiovasc Pharmacol. 2013;62(2):212–21.
Zahir H, Matsushima N, Halim AB, He L, Zhang G, Lee F, et al. Edoxaban administration following enoxaparin: a pharmacodynamic, pharmacokinetic, and tolerability assessment in human subjects. Thromb Haemost. 2012;108(1):166–75.
Mendell J, Lasseter K, Shi M. Effect of esomeprazole on edoxaban pharmacokinetics and pharmacodynamics when administered orally as tablet and as solution. Eur J. 2011;13 (Suppl 3):Abstract P1052.
Eriksson BI, Quinlan DJ, Eikelboom JW. Novel oral factor Xa and thrombin inhibitors in the management of thromboembolism. Annu Rev Med. 2011;62:41–57.
Song S, Kang D, Halim AB, Miller R. Population pharmacokinetic–pharmacodynamic modeling analysis of intrinsic FXa and bleeding from edoxaban treatment. J Clin Pharmacol. 2014;54(8):910–6.
Weitz JI, Connolly SJ, Patel I, Salazar D, Rohatagi S, Mendell J, et al. Randomised, parallel-group, multicentre, multinational phase 2 study comparing edoxaban, an oral factor Xa inhibitor, with warfarin for stroke prevention in patients with atrial fibrillation. Thromb Haemost. 2010;104(3):633–41.
Salazar DE, Mendell J, Kastrissios H, Green M, Carrothers TJ, Song S, et al. Modelling and simulation of edoxaban exposure and response relationships in patients with atrial fibrillation. Thromb Haemost. 2012;107(5):925–36.
Zafar MU, Vorchheimer DA, Gaztanaga J, Velez M, Yadegar D, Moreno PR, et al. Antithrombotic effects of factor Xa inhibition with DU-176b: phase-I study of an oral, direct factor Xa inhibitor using an ex-vivo flow chamber. Thromb Haemost. 2007;98(4):883–8.
Wolzt M, Samama MM, Kapiotis S, Ogata K, Mendell J, Kunitada S. Effect of edoxaban on markers of coagulation in venous and shed blood compared with fondaparinux. Thromb Haemost. 2011;105(6):1080–90.
Samama MM, Kunitada S, Oursin A, Depasse F, Heptinstall S. Comparison of a direct factor Xa inhibitor, edoxaban, with dalteparin and ximelagatran: a randomised controlled trial in healthy elderly adults. Thromb Res. 2010;126(4):e286–93.
Parasrampuria DA, Weilert D, Maa J-F, Dishy V, Kochan J, Shi M, et al. Switching from dabigatran or rivaroxaban to edoxaban: a biomarker assessment in healthy subjects. Thromb Haemost. 2014;12(Suppl 1): 1–106 (Abstract COA41).
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH harmonised tripartite guideline: the clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs E14. Geneva. 2005. Available at: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E14/E14_Guideline.pdf. Accessed 5 Nov 2015.
Mendell J, Basavapathruni R, Swearingen D, Draves A, Zhang G, Morganroth J. A thorough electrocardiogram study of edoxaban, a novel factor Xa inhibitor. J Clin Pharmacol. 2011;51(8):1241–6.
Mendell J, Noveck RJ, Shi M. A randomized trial of the safety, pharmacokinetics and pharmacodynamics of edoxaban, an oral factor Xa inhibitor, following a switch from warfarin. Br J Clin Pharmacol. 2013;75(4):966–78.
Ruff CT, Giugliano RP, Braunwald E, Mercuri M, Curt V, Betcher J, et al. Transition of patients from blinded study drug to open-label anticoagulation: the ENGAGE AF–TIMI 48 trial. J Am Coll Cardiol. 2014;64(6):576–84.
Acknowledgments
Dolly A. Parasrampuria was an employee of Daiichi Sankyo Pharma Development (Edison, NJ, USA) at the time when this manuscript was initiated. Kenneth E. Truitt is an employee of Daiichi Sankyo Pharma Development. Medical writing and editorial support was provided by Meryl Gersh, PhD, of AlphaBioCom, LLC (King of Prussia, PA, USA) and funded by Daiichi Sankyo (Parsippany, NJ, USA).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Parasrampuria, D.A., Truitt, K.E. Pharmacokinetics and Pharmacodynamics of Edoxaban, a Non-Vitamin K Antagonist Oral Anticoagulant that Inhibits Clotting Factor Xa. Clin Pharmacokinet 55, 641–655 (2016). https://doi.org/10.1007/s40262-015-0342-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-015-0342-7