
Abstract
Background
While in vitro and animal studies have shown reduced cytochrome P450 (CYP) 3A activity due to obesity, clinical studies in (morbidly) obese patients are scarce. As CYP3A activity may influence both clearance and oral bioavailability in a distinct manner, in this study the pharmacokinetics of the CYP3A substrate midazolam were evaluated after semi-simultaneous oral and intravenous administration in morbidly obese patients, and compared with healthy volunteers.
Methods
Twenty morbidly obese patients [mean body weight 144 kg (range 112–186 kg) and mean body mass index 47 kg/m2 (range 40–68 kg/m2)] participated in the study. All patients received a midazolam 7.5 mg oral and 5 mg intravenous dose (separated by 159 ± 67 min) and per patient 22 samples over 11 h were collected. Data from 12 healthy volunteers were available for a population pharmacokinetic analysis using NONMEM®.
Results
In the three-compartment model in which oral absorption was characterized by a transit absorption model, population mean clearance (relative standard error %) was similar [0.36 (4 %) L/min], while oral bioavailability was 60 % (13 %) in morbidly obese patients versus 28 % (7 %) in healthy volunteers (P < 0.001). Central and peripheral volumes of distribution increased substantially with body weight (both P < 0.001) and absorption rate (transit rate constant) was lower in morbidly obese patients [0.057 (5 %) vs. 0.130 (14 %) min–1, P < 0.001].
Conclusions
In morbidly obese patients, systemic clearance of midazolam is unchanged, while oral bioavailability is increased. Given the large increase in volumes of distribution, dose adaptations for intravenous midazolam should be considered. Further research should elucidate the exact physiological changes at intestinal and hepatic level contributing to these findings.
Similar content being viewed by others


No influence of obesity on the systemic clearance of the cytochrome P450 3A substrate midazolam could be identified when morbidly obese and non-obese subjects were studied. |
Midazolam oral bioavailability is increased in morbidly obese individuals. |
Midazolam volume of distribution increases substantially with body weight, necessitating dose adaptations for intravenous midazolam in morbidly obese patients. |
1 Introduction
The prevalence of obesity [body mass index (BMI) >30 kg/m2] and morbid obesity (BMI >40 kg/m2) is increasing rapidly. In 2010, 6.6 % (15.5 million) of the US adult population was morbidly obese, a 70 % increase since 2000 [1], while 36 % of the US population was obese [2]. In Europe, approximately 20 % of the adult population is currently obese [3].
In obese mice, studies have shown reduced hepatic cytochrome P450 (CYP) 3A protein expression [4, 5]. Similarly, in in vitro studies with hepatocytes from human fatty livers, reduced CYP3A expression and activity has been reported [6, 7] with increasing severity of fatty liver and non-alcoholic steatohepatitis (NASH), which are both highly associated with (morbid) obesity [8, 9]. However, these measurements concern absolute values and were not normalized for the weight of the whole liver and/or the body weight of the mouse. CYP3A is an important enzyme system that is responsible for the primary metabolism of 25 % of all clinically used drugs [10], including many drugs that are relevant for obese patients, such as statins (HMG-CoA reductase inhibitors), cardiovascular drugs, antipsychotics, and oncolytic drugs [11]. In obese compared with non-obese subjects, it was shown that hepatic and intestinal CYP3A protein expression decreased with increasing BMI [12] and that oral clearance (CL/F) of CYP3A substrates such as triazolam, carbamazepine, and taranabant was lower [13, 14], even though a similar systemic clearance (CL) was found in obese individuals for midazolam [15]. An explanation for the reduction in CYP3A activity upon obesity could be an increased state of inflammation caused by infiltration of macrophages and adipocytes into the adipose tissue excreting inflammation markers and adipokines, such as interleukin (IL)-6 and tumor necrosis factor (TNF)-α [16–18]. Both in vitro and animal studies have shown that inflammation factors such as IL-6 may decrease CYP3A expression, resulting in down-regulation of CYP3A-mediated metabolism [19–23]. Finally, reduced CYP3A activity due to inflammation has also been shown in critically ill patients [24, 25].
Midazolam, which is considered a specific marker of CYP3A activity because it is primarily metabolized by CYP3A [26], is a widely applied oral or intravenous drug for sleeping disorders, (pre)anesthesia, sedation for scopic interventions, and in the intensive care unit. As CYP3A is located in both the liver and the intestines, the activity of the CYP3A enzyme is an important determinant of midazolam CL and oral bioavailability [27, 28]. In view of the ever-increasing body weights of morbidly obese patients, in this pharmacokinetic study we evaluate the influence of morbid obesity on CYP3A-mediated CL and oral bioavailability of midazolam when studied after semi-simultaneous oral and intravenous administration, allowing these parameters to be characterized in a distinct manner. For the analysis, midazolam data from a healthy volunteer study with the same study design were also available. The results of this study are used to illustrate the consequences for dosing of oral and intravenous midazolam in morbidly obese patients.
2 Methods
2.1 Study Design and Patients
This prospective observational study in morbidly obese patients (NTC01519726 and EudraCT 2011-003293-93) was approved by the local human research and ethics committee of the Sint Antonius Ziekenhuis (VCMO, NL35861.100.11) and conducted in accordance with the principles of the Declaration of Helsinki and the Medical Research Involving Human Subjects Act (WMO) of The Netherlands. Before participation, all patients gave written informed consent.
Adult morbidly obese patients (BMI >40 kg/m2) undergoing laparoscopic gastric bypass or sleeve surgery were eligible for inclusion in the study. Patients were excluded if they used CYP3A-inducing or -inhibiting medication according to the Cytochrome P450 Drug Interaction Table [11], used products containing grapefruit, wild grape, banpeiyu, pomegranate, star fruit, or black berry within 2 weeks before the study, were pregnant or breastfeeding, or suffered from renal insufficiency (estimated glomerular filtration rate [Modification of Diet in Renal Disease (MDRD4) <60 mL/min].
2.2 Study Procedure
Twenty morbidly obese patients were studied on the day of laparoscopic bariatric surgery after an overnight fast. Midazolam was administered in a semi-simultaneous manner. Approximately 2.5 h before induction of anesthesia, patients received midazolam 7.5 mg orally as a tablet (Dormicum®, Roche). At the induction of anesthesia (159 ± 67 min after the oral dose), 5 mg of intravenous midazolam (Midazolam Actavis 5 mg/mL, Actavis) was administered. Blood samples were collected at T = 0, 5, 15, 30, 45, 55, 65, 75, 90, 120, and 150 min after the oral dose and T = 5, 15, 30, 30, 90, 120, 150, 180, 210, 270, 330, 390, and 510 min after the intravenous dose. Blood samples were collected in lithium heparin tubes and centrifuged at 3,000 rpm for 10 min at 4 °C. Plasma was separated and immediately stored at −80 °C until analysis.
Blood samples to measure markers of liver function [aspartate aminotransferase (AST), alanine aminotransferase (ALT), bilirubin, γ-glutamyltransferase, and albumin] in morbidly obese patients were collected before the oral midazolam dose was administered.
2.3 Control Group
Data from 12 healthy volunteers receiving midazolam in an identical semi-simultaneous dosing design were available for analysis (EudraCT 2009-010331-40). Subjects had to fast from 2 h before drug administration and received a 2 mg oral midazolam solution (Midazolam, Synthon) at 10:00 am. After 150 min they received a 1 mg intravenous midazolam dose (Midazolam, Synthon) and blood samples were collected at T = 15, 30, 45, 60, 65, 70, 75, 80, 90, 120, 148, 155, 165, 180, 210, 240, 270, 330, and 390 min after oral dose.
2.4 Drug Assay
In the plasma samples from morbidly obese patients, midazolam was analyzed using a MassTox® TDM series A BASIC-Kit for ultra performance liquid chromatography (UPLC)–tandem mass spectrometry (MS/MS) from Chromsystems Instruments and Chemicals GmbH (Gräfelfing/München, Germany), a commercially available kit including mobile phases, dilution buffers, and extraction buffer. For sample preparation, 25 μL of MassTox® BASIC-Kit A extraction buffer was added to 50 μL of the sample. After short vortex mixing, this mixture was let to incubate for 2 min. Then 250 μL MassTox® BASIC-Kit A internal standard mix was added and the mixture was vortexed for 30 s and centrifuged at 15,000 g for 5 min. Ten µL of the supernatant was injected onto the UPLC column (MasterColumn, Chromsystems) without a precolumn using a Waters Acquity UPLC system connected to a Waters TQD (TQ Detector) mass spectrometer with electrospray ionization. The column was kept at 25 °C. Eluent was used at a flow rate of 0.6 mL/min. Intra-assay and inter-assay coefficients of variation were 4.7 and 3.3 %, respectively. Midazolam recovery was 90 %. The lower limit of quantification (LLOQ) was 0.8 ng/mL.
In the plasma samples from healthy volunteers, midazolam was measured using a validated liquid chromatographic–tandem mass spectrometric (LC-MS/MS) assay. Briefly, 500 µL acetonitrile containing midazolam-D4 (4 µg/L) was added to 200 µL serum. After 3 min vortex mixing and 5 min centrifugation at ambient temperature the supernatant was collected and transferred into an autosampler vial. Next, 10 µL was injected on an Atlantis T3 C18 3 µm column (2.1 × 50 mm; Waters), protected with a guard column (ODS; 4 × 3 mm), which was kept at 30 °C. Gradient elution was performed with a mobile phase consisting of 0.1 % v/v aqueous formic acid and 0.1 % v/v formic acid in acetonitrile at a flow rate of 0.3 mL/min. The effluent was monitored with a Micromass Quattro Micro triple-quadrupole mass spectrometric detector (Waters). The detector was operated in the positive electrospray ionization mode and configured in the multiple reaction monitoring (MRM) mode. Within-day and between-day inaccuracy and imprecision were less than 5 %. The LLOQ was 0.3 ng/mL.
2.5 Population Pharmacokinetic Analysis and Internal Validation
Population pharmacokinetic modeling was performed on all data by means of non-linear mixed-effects modelling using NONMEM® (version 7.2; GloboMax LLC, Hanover, MD, USA) [29]. Pirana (2.7.1; Pirana Software & Consulting BV) and R (2.15) were used to visualize the data. All midazolam plasma concentration values that were received from the laboratory were inserted in the datafile, even if these were below the limit of quantification (LOQ). Of the 434 samples from morbidly obese patients, 42 were below the LOQ. For healthy volunteers, no data were below the LOQ.
Discrimination between different models was made by comparison of the objective function value (OFV; i.e., −2 log likelihood [−2LL]). A P value below 0.05, representing a decrease of 3.84 in the OFV for one degree of freedom, was considered statistically significant. In addition, goodness-of-fit plots (observed versus individual-predicted concentrations, observed versus population-predicted concentrations, conditional weighted residuals versus time, and conditional weighted residuals versus population-predicted concentrations plots) were used for diagnostic purposes. Furthermore, the confidence interval of the parameter estimates, the correlation matrix, and visual improvement of the individual plots were used to evaluate the model. The internal validity of the population pharmacokinetic model was assessed by the bootstrap re-sampling method using 500 replicates and normalized prediction distribution errors (NPDE) using 1,000 simulation of each dataset [30]. Parameters obtained with the bootstrap replicates were compared with the estimates obtained from the original dataset. NPDE plots were checked for normal distribution characteristics and trends in the data errors [30].
For the structural model, one-, two, and three-compartment models with an oral dosing compartment were tested. To describe the midazolam oral absorption phase, zero-order and first-order absorption models were tested, in addition to a lag time model and a transit absorption model [31]. For the transit absorption model, a varying number of transit compartments was tested. As the transit rate (Ktr) was set equal to the absorption rate (Ka), the mean transit time (MTT) can be calculated from Ktr with 1/(Ktr*(n + 1)), in which n is the number of transit compartments [32].
For the statistical model, the individual parameter estimate (empirical Bayes estimate or post hoc value) of the ith individual was modelled according to (Eq. 1):
where θ mean is the population mean and η i is a random variable for the ith individual with a mean of zero and variance of ω 2, assuming log-normal distribution in the population.
For residual variability, resulting from assay errors, model misspecifications, and other unexplained sources, a proportional error model and a combined proportional and additive model was tested for each of the datasets. The jth observed midazolam concentration of the ith healthy volunteer (Y ij ) is described by Eq. 2, while the jth observed midazolam concentration of the ith morbidly obese patient (Y ij ) is described by Eq. 3:
where C pred, ij is the population-predicted midazolam concentration of the ith individual at the jth time, and ε ij is a random variable with a mean of zero and variance of σ 2.
2.6 Covariate Analysis
Covariates were plotted independently against the individual post hoc values and eta estimates of pharmacokinetic parameters to visualize potential relations. The following covariates were tested: total body weight (TBW), BMI, lean body weight (LBW) [33], sex, morbid obesity and age. All covariates except for sex and morbid obesity were tested using linear and allometric equations (Eqs. 4 and 5):
where P i and P p represent individual and population parameter estimates, respectively; COV represents the covariate; COVmedian represents the median value of the covariate for the population; W represents a correlation factor between the population pharmacokinetic parameters and the change in covariate value; and X represents the exponential scaling factor for a power function. The binary covariates ‘sex’ and ‘morbid obesity’ were tested using Eq. 6:
where P i and P p represent the individual and population parameter estimate, and Z represents the estimated factor of increase or decrease for the patients subgroup with COV equalling 1.
Potential covariates were separately entered into the model and statistically tested by use of the OFV and, if applicable, the 95 % confidence interval of the additional parameter. In addition, if applicable, it was evaluated whether the inter-individual variability (eta) in the parameter concerned decreased upon inclusion of the covariate on the parameter and whether the trend in the eta versus covariate plot had resolved. When more than one significant covariate for the simple model was found, the covariate-adjusted model with the largest decrease in the OFV was chosen as a basis to sequentially explore the influence of additional covariates with the use of the same criteria. Finally, after forward inclusion (P < 0.05), a backward exclusion procedure was applied to justify the inclusion of a covariate (P < 0.01). The choice of the covariate model was further evaluated as discussed in the Population Pharmacokinetic Analysis and Internal Validation section.
2.7 Model Simulations
Using NONMEM® 7.2, the final population pharmacokinetic model was used to simulate the concentration–time profiles of four typical patients from the dataset, including one healthy volunteer of 76 kg and three morbidly obese patients of 112, 145, and 186 kg. The 76 and 145 kg dose simulations represent the median body weight of the healthy volunteer and morbidly obese patient group, respectively. In addition, the 112 and 186 kg dose simulations represent the extremes of the body weight range of the morbidly obese patient group (see Table 1).
3 Results
3.1 Patients and Data
Twenty morbidly obese patients participated in this study and a mean of 22 ± 3 samples per patient were available for analysis. In addition, data from 12 healthy volunteers with 19 midazolam concentrations per subject were used as a control group in this analysis.
Liver function markers in morbidly obese subjects were all within three times the upper limit of normal, with the vast majority being within two times the upper limit of normal of the different markers. The demographics of all subjects are summarized in Table 1.
3.2 Population Pharmacokinetic Model and Validation
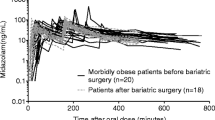
A three-compartment model with two equalized peripheral volumes of distribution best fitted the data. This model showed an improved fit over a two-compartment model, while a full three-compartment model could not be estimated with adequate precision. The pharmacokinetic model was parameterized in terms of oral Ka, oral bioavailability, volume of distribution of the central compartment (V1), two equalized volumes of distribution of the peripheral compartments (V2 and V3), inter-compartmental clearances from the central compartment to each peripheral compartment (Q and Q2), and clearance from the central compartment (CL). Midazolam oral absorption described by three transit absorption compartments proved superior over the other oral absorption models (zero and/or first-order absorption models or a lag time model) (Fig. 1). An omega block was implemented to account for the correlation between the central and peripheral volume of distribution. Table 2 shows the parameter estimates of the simple model without covariates.

Schematic illustration of the population pharmacokinetic model of oral and intravenous administered midazolam. CL clearance, F oral bioavailability, i.v. intravenous, Ka oral absorption rate, Ktr transit rate constant, mdz midazolam, Q inter-compartmental clearance to first peripheral volume, Q2 inter-compartmental clearance to second peripheral volume, V volume of distribution
In the covariate analysis, a significant influence of TBW or ‘morbid obesity’ was found on four different parameters, which is visualized in Fig. 2 where the post hoc parameters of the simple model without covariates are given. It was found that the peripheral volumes of distribution increased in a non-linear manner with TBW (P < 0.001, −24 ΔOFV), and that the central volume of distribution showed a linear increase with body weight (P < 0.001, −17 ΔOFV). For oral bioavailability and Ka (or Ktr), ‘morbid obesity’ was a significant covariate and significantly improved the model (P < 0.001, −22 ΔOFV and P < 0.001, −20 ΔOFV, respectively). For clearance, there was a trend towards a positive influence of LBW but not for TBW; however, the statistical significance was insufficient for inclusion of LBW in the final covariate model (P < 0.05, −4 ΔOFV), the estimated correlation factor was not estimated with adequate precision, and the eta for clearance value was not reduced. Eta distributions for clearance versus body weight and LBW of the simple and final covariate model are included in the Electronic Supplementary Material (ESM 1). After inclusion of the covariates in the model, the trends in eta value of the parameter and the covariate had disappeared and no residual trends were observed (Electronic Supplementary Material, ESM 2). All parameter estimates of the final model are shown in Table 2. Figure 3 demonstrates the goodness-of-fit plots of the final covariate model. The plots show no remaining bias for predicted midazolam concentrations in morbidly obese patients or healthy volunteers. The final model was internally validated by means of 500 bootstrap runs (Table 2), which were successful in 96 % of the runs and confirmed the parameter values. Finally, an NPDE analysis was performed showing a normal distribution of errors without trends, except for the initial midazolam concentrations in morbidly obese patients, which were below the LOQ (see Electronic Supplementary Material, ESM 3). The NONMEM® control file of the final model is provided as a Electronic Supplementary Material (ESM 4)

Post hoc parameter estimates of morbidly obese individuals (n = 20, black dots) and healthy individuals (n = 12, gray dots) from the simple pharmacokinetic model versus total body weight, including central volume of distribution versus total body weight (a), peripheral volume of distribution versus total body weight (b), oral bioavailability versus total body weight (c), and oral absorption rate versus total body weight (d)

Observed versus individual predicted midazolam concentrations (a), observed versus population predicted midazolam concentrations (b), conditional weighted residuals versus population predicted midazolam concentrations (c), and conditional weighted residuals versus time of the final model for 20 morbidly obese patients (black dots) and 12 healthy volunteers (gray dots). The dashed line represents the line of identity (x = y). CWRES conditional weighted residuals
3.3 Simulations
Figure 4 shows population predicted midazolam concentrations after a 5 mg intravenous bolus dose, a 2.5 mg/h continuous infusion for a duration of 10 days (14,400 min), and a 7.5 mg oral dose in four typical subjects from the dataset (i.e., 76, 112, 145, and 186 kg). The plot shows that for the intravenous bolus dose, midazolam maximum (peak) concentrations (C max) are lower in morbidly obese patients (Fig. 4a), which may be the result of the higher central volume of distribution in morbidly obese patients. In addition, the plot illustrates the longer midazolam elimination half-life (t ½) in morbidly obese patients, which can be attributed to the increase in volumes of distribution with body weight, while clearance is the same in all patients. The continuous intravenous infusion simulation (Fig. 4b) shows that with increasing body weight the midazolam steady-state concentrations are reached at a later timepoint. In a 76 kg healthy volunteer, steady state is expected to be reached after 24 h, while this is 170 h in a 145 kg morbidly obese patient and >240 h in a 186 kg morbidly obese patient (Fig. 4b). Finally, the oral midazolam dose simulations (Fig. 4c) show that the time to reach the C max is later (31 min) in morbidly obese patients than in healthy volunteers, while the C max is slightly lower in morbidly obese patients.

Population predicted midazolam concentrations over time in three typical morbidly obese patients (112, 145, and 186 kg) and one healthy volunteer (76 kg) after a 5 mg intravenous bolus dose (logarithmic scale) (a), a 2.5 mg/h continuous infusion (linear scale) (b), and a 7.5 mg oral dose (linear scale) (c)
4 Discussion
As there is only limited information on the influence of morbid obesity on CYP3A-mediated clearance of drugs in patients, this study aimed to evaluate the pharmacokinetics of the CYP3A substrate midazolam in morbidly obese patients following oral and intravenous administration. As clearance after oral dosing is dependent on oral bioavailability, which may be influenced by CYP3A enzyme activity in the intestines, this semi-simultaneous design allows for an estimation of both CL and oral bioavailability in a distinct manner. An available dataset of midazolam concentrations collected on the basis of an equivalent study design in healthy volunteers allowed for a head-to-head comparison between morbidly obese patients and non-obese healthy subjects. The results from this study show that midazolam clearance was similar in morbidly obese patients and healthy volunteers, oral bioavailability was substantially higher (60 % instead of 28 %), oral Ka was reduced, and that the central and peripheral volumes of distribution increased substantially with body weight. Particularly for intravenous dosing, the net results of all these changes should be considered when administering midazolam to morbidly obese patients.
In this study, we could not identify an influence of morbid obesity on the CL of midazolam, even though a wide range in body weights was included in this study. We did find a trend of increasing midazolam clearance with LBW; however, this trend was not strong enough for inclusion in the final covariate model. Possibly, the patient numbers in this analysis (n = 12 + n = 20) are insufficient to adequately detect a small increase in clearance with LBW. While these results indicate a lack of change in absolute hepatic CYP3A-mediated metabolism of midazolam in morbidly obese individuals, the results are in contrast with our expectations of a lower midazolam clearance in morbidly obese patients which was based on reports in in vitro and animal studies [4–7] and on oral clearance of CYP3A substrates in studies in obese subjects [13, 14]. Assuming that indeed the relative CYP3A activity per unit of liver is reduced in morbidly obese patients [12], we hypothesize that this effect may be counteracted by a higher liver volume [34], resulting in a similar absolute hepatic CYP3A metabolizing capacity in both groups. In agreement with this hypothesis, Greenblatt et al. [15] also found no significant difference in absolute CL of midazolam between normal weight (66 ± 2 kg) and obese subjects (117 ± 8 kg) (0.53 ± 0.04 vs. 0.47 ± 0.04 L/min, respectively). However, for a study with triazolam, another benzodiazepine CYP3A substrate, a lower apparent total clearance of the drug from plasma after oral administration (CL/F) was found for obese patients [35]. Lower CL/F values were also found for obese patients for the CYP3A substrates taranabant and carbamazepine [36, 37]. Based on the results found in the current midazolam study upon both oral and intravenous administration, it may be hypothesized that these lower CL/F values [35–37] are due to an increase in oral bioavailability instead of a decrease in CL.
Oral bioavailability was found to be higher in morbidly obese patients than in healthy volunteers [0.60 (13.2 %) versus 0.28 (7.0 %)]. In contrast, Greenblatt et al. [15] found similar values of oral midazolam bioavailability in obese (0.42 ± 0.04) and normal weight patients (0.40 ± 0.03) (P > 0.05). This disagreement in results may be explained by the higher body weights of the morbidly obese subjects in our study versus the study of Greenblatt et al. [15] (mean of 144 ± 22 vs. 117 ± 8 kg). In addition, the concentration–time profiles after oral and intravenous midazolam of a non-obese and an obese subject shown in their publication may also point at a higher bioavailability in the obese patient. We anticipate that the increase in oral bioavailability in morbidly obese patients found in our study may be due to reduced CYP3A-metabolizing activity in the intestines. Ulvestad et al. [12] found in a study with 19 obese individuals [median BMI 45 (34–59) kg/m2] that CYP3A4 protein expression in the small intestine and liver is lower with increasing BMI. Another possible cause of increased bioavailability is an increase in splanchnic blood flow, which has been reported before in morbidly obese patients. An increase in villous blood flow in the gut wall will cause an increase in substrate transport and thus carry the substrate away from the intestinal CYP3A-metabolizing enzymes [38, 39]. Moreover, increased intestinal permeability may be responsible for increased midazolam bioavailability as obese patients showed increased paracellular absorption measured with lactulose and chromium (Cr)-EDTA, which may possibly be due to reduced tight junction function [40, 41]. A question would be whether the observed difference in Ka between the morbidly obese patients and healthy volunteers, which can be attributed to a difference in formulation (tablet vs. oral solution), may have contributed to the reported difference in oral bioavailability. In our opinion, this difference in formulation is unlikely to influence oral bioavailability, as midazolam is a highly soluble and permeable drug that is expected to be 100 % absorbed in the intestines [42]. Correspondingly, a study in which six healthy volunteers received a midazolam 10 mg oral solution, a 10 mg tablet, and a 5 mg intravenous bolus dose showed similar oral bioavailability after both oral dose formulations, 0.35 ± 0.07 versus 0.38 ± 0.12 (P > 0.6), indicating no influence of oral formulation on midazolam bioavailability [43].
To understand the net result of the influence of different degrees of (morbid) obesity on each of the midazolam pharmacokinetic parameters, simulations using the final model were performed to yield midazolam concentration–time profiles for subjects with different body weights. The dose simulations show that the same intravenous bolus dose to all subjects leads to lower initial concentrations in morbidly obese patients due to a substantially higher central and peripheral volume of distribution. This observed increase in volume of distribution is in agreement with the midazolam study of Greenblatt et al. [15] in which a substantial increase in total volume of distribution for obese versus normal weight subjects of 311 ± 27 versus 114 ± 7 L (P < 0.001) was also reported. Potentially, these results can be explained by an increase in body volume in terms of both well-perfused compartments (organ and blood volume) and adipose tissue with obesity, which is of specific relevance because midazolam is a lipophilic drug [44, 45]. As such, directly after an intravenous bolus dose, lower midazolam concentrations and associated effects may be expected in morbidly obese patients. In addition, morbidly obese patients show an increased t ½ (Fig. 4), which can be attributed to the larger volumes of distribution as well, as they allow for significant midazolam disposition from the blood and may lead to prolonged midazolam effects in morbidly obese patients versus healthy volunteers. In contrast, a similar midazolam oral dose will result in only slightly lower initial concentrations in morbidly obese patients versus healthy volunteers because the increased oral bioavailability counteracts the influence of increased central volume of distribution on midazolam C max values (Fig. 4). Finally, the increase in the volumes of distribution with body weight also explains the increased duration for morbidly obese patients to reach steady-state concentrations after a continuous intravenous infusion. This phenomenon has been described before for diazepam in obese patients [46]. Therefore, a loading dose or a higher initial continuous infusion rate may be considered to reach midazolam steady-state concentrations more rapidly in morbidly obese patients.
There are some limitations to this study. Firstly, the sampling duration after oral administration may have been relatively short. Particularly in morbidly obese patients, the midazolam C max values after the oral dose occurred at approximately 90 min post-dose, leaving only a 60 min time interval to collect data on the concentration decline after the oral dose before the intravenous dose was administered. However, in six of the 20 patients this interval was >180 min (due to a delay in the surgery schedule), thus providing significant information on the midazolam pharmacokinetics after oral absorption in the morbidly obese patients. Secondly, the healthy volunteer group lacks a late sample post-intravenous dose, which have may have an effect on the clearance and peripheral volume of distribution estimates of the healthy volunteers and thus obscure the covariate analysis. However, estimated pharmacokinetic parameter values for this group closely match those found in previous midazolam pharmacokinetic studies in healthy volunteers, indicating adequate precision of the pharmacokinetic parameters in healthy volunteers and justifying the results from the covariate analysis. Thirdly, morbidly obese patients underwent surgery during the study, which may influence midazolam clearance and distribution. However, we think that surgery was only of minor influence as only the intravenous dose was administered during surgery and CL and volume of distribution found in this study were fairly similar to earlier reported values in non-surgery obese patients [15]. Finally, the stable isotope method for determining oral bioavailability in a single person on a single occasion may have been preferable over the current semi-simultaneous dosing design, though the semi-simultaneous oral—intravenous administration method has proved a reliable and accurate method for estimating oral bioavailability and CL in a single person on a single occasion as well [47–50]. Moreover, the available control data (midazolam concentrations in healthy volunteers) was gathered in a semi-simultaneous design. Lastly, the preparation of the labeled drug and the determination of the labeled drug in the samples is very expensive and labor intensive. For these reasons, we have chosen to apply the semi-simultaneous design.
5 Conclusion
This study shows that midazolam hepatic clearance was not changed in morbidly obese patients versus healthy volunteers, while oral bioavailability was increased in morbidly obese patients. Midazolam central and peripheral volumes of distribution increased substantially with body weight, resulting in lower midazolam concentrations after intravenous bolus administration and in an increased duration to reach steady-state concentrations after midazolam continuous infusion in morbidly obese patients in comparison with healthy volunteers. Finally, initial midazolam concentrations after an oral dose were similar in morbidly obese patients versus healthy volunteers. Further research should elucidate the exact physiological changes at the intestinal and hepatic level contributing to these findings.
References
Sturm R, Hattori A. Morbid obesity rates continue to rise rapidly in the United States. Int J Obes (Lond). 2013;37(6):889–91. doi:10.1038/ijo.2012.159.
Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of Obesity in the United Stated, 2009–2010. NCHS Data Brief 2012. http://www.cdc.gov/nchs/data/databriefs/db82.pdf. Accessed 11 Aug 2014.
World Obesity Federation. 2014. Registered in England and Wales, 3802726. http://www.worldobesity.org/aboutobesity/world-map-obesity/. Accessed 15 Aug 2014.
Yoshinari K, Takagi S, Yoshimasa T, Sugatani J, Miwa M. Hepatic CYP3A expression is attenuated in obese mice fed a high-fat diet. Pharm Res. 2006;23(6):1188–200. doi:10.1007/s11095-006-0071-6.
Ghose R, Omoluabi O, Gandhi A, Shah P, Strohacker K, Carpenter KC, et al. Role of high-fat diet in regulation of gene expression of drug metabolizing enzymes and transporters. Life Sci. 2011;89(1–2):57–64. doi:10.1016/j.lfs.2011.05.005.
Fisher CD, Lickteig AJ, Augustine LM, Ranger-Moore J, Jackson JP, Ferguson SS, et al. Hepatic cytochrome P450 enzyme alterations in humans with progressive stages of nonalcoholic fatty liver disease. Drug Metab Dispos. 2009;37(10):2087–94. doi:10.1124/dmd.109.027466.
Kolwankar D, Vuppalanchi R, Ethell B, Jones DR, Wrighton SA, Hall SD, et al. Association between nonalcoholic hepatic steatosis and hepatic cytochrome P-450 3A activity. Clin Gastroenterol Hepatol. 2007;5(3):388–93. doi:10.1016/j.cgh.2006.12.021.
Wree A, Kahraman A, Gerken G, Canbay A. Obesity affects the liver—the link between adipocytes and hepatocytes. Digestion. 2011;83(1–2):124–33. doi:10.1159/000318741.
Moretto M, Kupski C, Mottin CC, Repetto G, Garcia Toneto M, Rizzolli J, et al. Hepatic steatosis in patients undergoing bariatric surgery and its relationship to body mass index and co-morbidities. Obes Surg. 2003;13(4):622–4.
Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther. 2013;138(1):103–41. doi:10.1016/j.pharmthera.2012.12.007.
Flockhart A. Cytochrome P450 drug interaction table. Indiana University School of Medicine. http://medicine.iupui.edu/clinpharm/ddis/clinical-table/. Accessed 29 Apr 2014.
Ulvestad M, Skottheim IB, Jakobsen GS, Bremer S, Molden E, Asberg A, et al. Impact of OATP1B1, MDR1, and CYP3A4 expression in liver and intestine on interpatient pharmacokinetic variability of atorvastatin in obese subjects. Clin Pharmacol Ther. 2013;93(3):275–82. doi:10.1038/clpt.2012.261.
Kotlyar M, Carson SW. Effects of obesity on the cytochrome P450 enzyme system. Int J Clin Pharmacol Ther. 1999;37(1):8–19.
Brill MJ, Diepstraten J, van Rongen A, van Kralingen S, van den Anker JN, Knibbe CA. Impact of obesity on drug metabolism and elimination in adults and children. Clin Pharmacokinet. 2012;51(5):277–304. doi:10.2165/11599410-000000000-00000.
Greenblatt DJ, Abernethy DR, Locniskar A, Harmatz JS, Limjuco RA, Shader RI. Effect of age, gender, and obesity on midazolam kinetics. Anesthesiology. 1984;61(1):27–35.
Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115(5):1111–9. doi:10.1172/JCI25102.
Fantuzzi G. Adipose tissue, adipokines, and inflammation. J Allergy Clin Immunol. 2005;115(5):911–9. doi:10.1016/j.jaci.2005.02.023 quiz 20.
Mathieu P, Lemieux I, Despres JP. Obesity, inflammation, and cardiovascular risk. Clin Pharmacol Ther. 2010;87(4):407–16. doi:10.1038/clpt.2009.311.
Jover R, Bort R, Gomez-Lechon MJ, Castell JV. Down-regulation of human CYP3A4 by the inflammatory signal interleukin-6: molecular mechanism and transcription factors involved. FASEB J. 2002;16(13):1799–801. doi:10.1096/fj.02-0195fje.
Aitken AE, Richardson TA, Morgan ET. Regulation of drug-metabolizing enzymes and transporters in inflammation. Annu Rev Pharmacol Toxicol. 2006;46:123–49. doi:10.1146/annurev.pharmtox.46.120604.141059.
Renton KW. Cytochrome P450 regulation and drug biotransformation during inflammation and infection. Curr Drug Metab. 2004;5(3):235–43.
Charles KA, Rivory LP, Brown SL, Liddle C, Clarke SJ, Robertson GR. Transcriptional repression of hepatic cytochrome P450 3A4 gene in the presence of cancer. Clin Cancer Res. 2006;12(24):7492–7. doi:10.1158/1078-0432.CCR-06-0023.
Yang J, Hao C, Yang D, Shi D, Song X, Luan X, et al. Pregnane X receptor is required for interleukin-6-mediated down-regulation of cytochrome P450 3A4 in human hepatocytes. Toxicol Lett. 2010;197(3):219–26. doi:10.1016/j.toxlet.2010.06.003.
Vet NJ, de Hoog M, Tibboel D, de Wildt SN. The effect of critical illness and inflammation on midazolam therapy in children. Pediatr Crit Care Med. 2010. doi:10.1097/PCC.0b013e3181fe406d.
Shelly MP, Mendel L, Park GR. Failure of critically ill patients to metabolise midazolam. Anaesthesia. 1987;42(6):619–26.
Streetman DS, Bertino JS Jr, Nafziger AN. Phenotyping of drug-metabolizing enzymes in adults: a review of in-vivo cytochrome P450 phenotyping probes. Pharmacogenetics. 2000;10(3):187–216.
Lindell M, Karlsson MO, Lennernas H, Pahlman L, Lang MA. Variable expression of CYP and Pgp genes in the human small intestine. Eur J Clin Invest. 2003;33(6):493–9.
Thorn M, Finnstrom N, Lundgren S, Rane A, Loof L. Cytochromes P450 and MDR1 mRNA expression along the human gastrointestinal tract. Br J Clin Pharmacol. 2005;60(1):54–60. doi:10.1111/j.1365-2125.2005.02389.x.
Beal S, Sheiner LB, Boeckmann A. NONMEM user’s guides (1989–2006). Ellicott City: Icon Development Solutions; 2006.
Brendel K, Comets E, Laffont CM, Laveille C, Mentré F. Metrics for external model evaluation with an application to the population pharmacokinetics of gliclazide. Pharm Res. 2006;23:2036–49.
Savic RM, Jonker DM, Kerbusch T, Karlsson MO. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J Pharmacokinet Pharmacodyn. 2007;34(5):711–26. doi:10.1007/s10928-007-9066-0.
Bonate P. Pharmacokinetic-pharmacodynamic modeling and simulation. New York: Springer; 2006.
Janmahasatian S, Duffull SB, Ash S, Ward LC, Byrne NM, Green B. Quantification of lean bodyweight. Clin Pharmacokinet. 2005;44(10):1051–65.
Lewis MC, Phillips ML, Slavotinek JP, Kow L, Thompson CH, Toouli J. Change in liver size and fat content after treatment with Optifast very low calorie diet. Obes Surg. 2006;16(6):697–701. doi:10.1381/096089206777346682.
Abernethy DR, Greenblatt DJ, Divoll M, Smith RB, Shader RI. The influence of obesity on the pharmacokinetics of oral alprazolam and triazolam. Clin Pharmacokinet. 1984;9(2):177–83.
Li XS, Nielsen J, Cirincione B, Li H, Addy C, Wagner J, et al. Development of a population pharmacokinetic model for taranabant, a cannibinoid-1 receptor inverse agonist. AAPS J. 2010;12(4):537–47. doi:10.1208/s12248-010-9212-2.
Caraco Y, Zylber-Katz E, Berry EM, Levy M. Significant weight reduction in obese subjects enhances carbamazepine elimination. Clin Pharmacol Ther. 1992;51(5):501–6.
Yang J, Jamei M, Yeo KR, Tucker GT, Rostami-Hodjegan A. Prediction of intestinal first-pass drug metabolism. Curr Drug Metab. 2007;8(7):676–84.
Rostami-Hodjegan A, Tucker GT. The effects of portal shunts on intestinal cytochrome P450 3A activity. Hepatology. 2002;35(6):1549–50. doi:10.1053/jhep.2002.33215; author reply 50–1.
Teixeira TF, Souza NC, Chiarello PG, Franceschini SC, Bressan J, Ferreira CL, et al. Intestinal permeability parameters in obese patients are correlated with metabolic syndrome risk factors. Clin Nutr. 2012;31(5):735–40. doi:10.1016/j.clnu.2012.02.009.
Horton F, Wright J, Smith L, Hinton PJ, Robertson MD. Increased intestinal permeability to oral chromium (Cr)—EDTA in human Type 2 diabetes. Diabet Med. 2013. doi:10.1111/dme.12360.
Jamei M, Turner D, Yang J, Neuhoff S, Polak S, Rostami-Hodjegan A, et al. Population-based mechanistic prediction of oral drug absorption. AAPS J. 2009;11(2):225–37. doi:10.1208/s12248-009-9099-y.
Smith MT, Eadie MJ, Brophy TO. The pharmacokinetics of midazolam in man. Eur J Clin Pharmacol. 1981;19(4):271–8.
Alexander JK, Dennis EW, Smith WG, Amad KH, Duncan WC, Austin RC. Blood volume, cardiac output, and distribution of systemic blood flow in extreme obesity. Cardiovasc Res Cent Bull. 1962;1:39–44.
Messerli FH, Sundgaard-Riise K, Reisin E, Dreslinski G, Dunn FG, Frohlich E. Disparate cardiovascular effects of obesity and arterial hypertension. Am J Med. 1983;74(5):808–12.
Abernethy DR, Greenblatt DJ, Divoll M, Shader RI. Prolonged accumulation of diazepam in obesity. J Clin Pharmacol. 1983;23(8–9):369–76.
Karlsson MO, Bredberg U. Estimation of bioavailability on a single occasion after semisimultaneous drug administration. Pharm Res. 1989;6(9):817–21.
Karlsson MO, Bredberg U. Bioavailability estimation by semisimultaneous drug administration: a Monte Carlo simulation study. J Pharmacokinet Biopharm. 1990;18(2):103–20.
Bredberg U, Karlsson MO, Borgstrom L. A comparison between the semisimultaneous and the stable isotope techniques for bioavailability estimation of terbutaline in humans. Clin Pharmacol Ther. 1992;52(3):239–48.
Lee JI, Chaves-Gnecco D, Amico JA, Kroboth PD, Wilson JW, Frye RF. Application of semisimultaneous midazolam administration for hepatic and intestinal cytochrome P450 3A phenotyping. Clin Pharmacol Ther. 2002;72(6):718–28. doi:10.1067/mcp.2002.129068.
Acknowledgments
This study was sponsored by ZonMW (The Netherlands Organisation for Health Research and Development), project number 836011008. In addition, we thank bariatric nurses Brigitte Bliemer and Silvia Samsom for their help in recruiting patients. Margreke J. E. Brill, Anne van Rongen, Aletta P. I. Houwink, Jacobus Burggraaf, Bert van Ramshorst, René J. Wiezer, Eric P. A. van Dongen and Catherijne A. J. Knibbe have no potential conflicts of interest to declare.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
40262_2014_166_MOESM1_ESM.eps
ESM 1 Eta distributions for clearance for the simple (a and c) and final (b and d) pharmacokinetic model versus lean body weight (a and b) and body weight (c and d) in 20 morbidly obese patients (black dots) and 12 healthy volunteers (grey dots). Supplementary material 1 (EPS 199 kb)
40262_2014_166_MOESM2_ESM.eps
ESM 2 Eta distributions for central volume of distribution (a and b), peripheral volume of distribution (c and d), oral bioavailability (e and f), oral absorption rate (Ktr) (g and h) in 20 morbidly obese patients (black dots) and 12 healthy volunteers (grey dots) for the simple (a, c, e, g) and final midazolam pharmacokinetic model (b, d, f, h). Supplementary material 2 (EPS 390 kb)
40262_2014_166_MOESM3_ESM.eps
ESM 3 Normalized prediction distribution errors (npde) for the midazolam data from 20 morbidly obese patients (a) and from 12 healthy volunteers (b). The X indicates the time after dose and Y indicates the midazolam concentration in nmol/L. Supplementary material 3 (EPS 1117 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Brill, M.J.E., van Rongen, A., Houwink, A.P.I. et al. Midazolam Pharmacokinetics in Morbidly Obese Patients Following Semi-Simultaneous Oral and Intravenous Administration: A Comparison with Healthy Volunteers. Clin Pharmacokinet 53, 931–941 (2014). https://doi.org/10.1007/s40262-014-0166-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-014-0166-x