
Abstract
Background and Objectives
There is a clinical need for a liquid formulation of atomoxetine. We assessed the safety and bioequivalence of an atomoxetine oral solution.
Methods
This was an open-label, randomized, crossover study. Healthy adult male Japanese subjects (n = 42) with a cytochrome P450 2D6 extensive (including intermediate and ultrarapid) metabolizer genotype were administered atomoxetine 50 mg as oral solution and capsules once each, with a washout period >5 days between doses. Blood samples were used to analyze pharmacokinetic parameters, particularly maximum observed drug concentration (C max) and area under the concentration vs. time curve from time zero to the last time point with a measurable concentration (AUC0-last). Bioequivalence was concluded if the 90 % confidence interval of the ratio of geometric means between formulations for both C max and AUC0-last were within the interval of 0.8–1.25. Safety assessments included determination of adverse events. Taste was evaluated via a five-item questionnaire immediately and 10 min after taking atomoxetine oral solution.
Results
Forty subjects completed the study. Plasma concentration–time profiles of atomoxetine oral solution and capsules were similar, and the statistical analysis of systemic exposure showed that the two formulations were bioequivalent. Adverse events were mild and similar in type and frequency between the formulations. For taste acceptability, only 7.1 % of subjects responded that the oral solution would be difficult to take every day.
Conclusion
Atomoxetine oral solution is bioequivalent to atomoxetine capsules and potentially fulfills the need for an oral solution atomoxetine formulation that will facilitate treatment of children with attention-deficit hyperactivity disorder.
Similar content being viewed by others


Many children find swallowing tablets or capsules difficult, and easy-to-administer pediatric formulations of attention-deficit hyperactivity disorder (ADHD) medications are a recognized clinical need, especially considering the age of young ADHD patients. |
This study in healthy Japanese male subjects demonstrates that a newly developed oral solution of atomoxetine is bioequivalent to atomoxetine capsules with a comparable safety profile. |
Further, atomoxetine oral solution has an acceptable taste and palatability profile, which is critical for this type of formulation. |
1 Introduction
Atomoxetine hydrochloride is a selective norepinephrine reuptake inhibitor approved for the treatment of attention-deficit hyperactivity disorder (ADHD) in children aged 6 years and older, adolescents, and adults. Atomoxetine is licensed in many countries and regions, including USA, Europe, and Japan. Numerous randomized and open-label studies have shown that atomoxetine leads to significant short-and long-term improvement in ADHD symptoms and relevant rating scales in children [1–6] and adults [7, 8]. Atomoxetine has also been associated with improvements in quality of life, behavior, coping abilities, and functioning among children and young adults [9–12].
During the investigation of new medications, the development and use of age-appropriate pediatric formulations has been recommended by agencies such as the US Food and Drug Administration and the European Medicines Agency [13, 14]. The need for age-appropriate pediatric formulations that are also easy to administer is well recognized in clinical practice [15, 16] and has been demonstrated in children with ADHD. Specifically, a survey of ADHD specialists in Japan found that the development of a “child friendly” atomoxetine formulation was an important clinical need [17]. In the case of pediatric formulations, ease of administration relates to numerous factors, including ease of swallowing, palatability, and dose flexibility, to accommodate the wide range of physical sizes and weights of patients in a pediatric population [16, 18, 19].
Based on this clinical need, an oral solution of atomoxetine has been developed to provide a convenient, flexible, and stable dosage form, with acceptable palatability for patients who have difficulty swallowing capsules. To mask the specific taste of atomoxetine, sweeteners and raspberry flavor were included in the formulation. To add a new formulation, demonstration of bioequivalence between this newly developed oral solution formulation and the currently available capsule formulation is required to meet regulatory requirements [20]. Accordingly, the primary aim of this bioequivalence study was to compare the pharmacokinetic profile of atomoxetine oral solution with commercially available atomoxetine capsules in healthy adult subjects. The secondary aim of this study was to determine the safety profile of atomoxetine oral solution and atomoxetine capsules in the same population. Finally, an exploratory taste assessment of atomoxetine oral solution was conducted.
2 Methods
2.1 Study Design
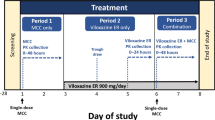
This open-label, randomized, crossover study in healthy adult male Japanese subjects was conducted at one study center in Fukuoka, Japan. Each subject was administered a single 50-mg dose of atomoxetine oral solution (Strattera Oral Solution®; Eli Lilly and Company, Indianapolis, IN, USA; 12.5 mL of 4 mg/mL) and atomoxetine capsules (Strattera®; Eli Lilly and Company, Indianapolis, IN, USA; two 25-mg capsules, long span = 15.8 mm; short span = 5.85 mm) each in a crossover fashion. Capsules were taken with approximately 200 mL of water. Subjects randomized to Cohort 1 received atomoxetine capsules and then, following a washout period of at least 5 days, received atomoxetine oral solution. Similarly, subjects randomized to Cohort 2 received atomoxetine oral solution and then, following a washout period of at least 5 days, received atomoxetine capsules.
For both the atomoxetine oral solution and atomoxetine capsules, each dose of atomoxetine was administered in the morning after an overnight fast (>10 h). Subjects then continued to fast an additional 4 h, followed by a meal. Water consumption was allowed, except 1 h prior to and 1 h after dosing. A regular diet was served to subjects during the study.
Blood samples were taken before each dose and at specified time points (0.25, 0.5, 0.75, 1, 1.25, 1.5, 2, 4, 6, 8, 12, 18, and 24 h) after each dose for analysis of pharmacokinetic parameters to assess bioequivalence. Safety assessments were performed for both formulations during the treatment periods and at a follow-up visit 5–9 days after the last dose. An exploratory taste evaluation was also performed for the oral solution.
The study was designed in accordance with the Japanese Guideline for Bioequivalence Studies for Generic Products [21] and was conducted in accordance with Good Clinical Practice and the 1964 Helsinki Declaration and its later amendments. Approval was granted by an ethical review board and all subjects provided written informed consent to participate in the study, including genotyping. The study was registered at www.clinicaltrials.gov (NCT01177943).
2.2 Study Subjects
Healthy male subjects aged 20–55 years with a body mass index of 17.6–26.4 kg/m2 were considered suitable for inclusion. Subjects categorized with a cytochrome P450 2D6 (CYP2D6) extensive metabolizer genotype, including ultrarapid metabolizers and intermediate metabolizers, were enrolled. (Blood samples for genotyping were collected at screening. Genotyping was performed by Beckman Coulter Genomics, Inc., Morrisville, NC, USA.) The extensive metabolizer genotype was defined as homozygous for the normal-functioning alleles, the poor metabolizer genotype as homozygous for null alleles, the ultrarapid metabolizer genotype as having more than three normal-functioning alleles, and the intermediate metabolizer genotype as having other allele combinations (normal function/decreased function, normal function/null, decreased function/decreased function, and decreased function/null). The following alleles were evaluated: *1, *2, and *35 as normal function; *9, *10, *17, *29, and *41 as decreased function; and *3, *4, *5, *6, *7, *8, *11, *12, *14/*14A, *15, *19, *20, *21, *36, and *40 as null.
2.3 Outcome Measures
Bioequivalence of the oral solution and capsule formulations was assessed via analysis of the following key pharmacokinetic parameters for the parent compound: maximum observed drug concentration (C max) and area under the concentration versus time curve (AUC) from time zero to the last time point with a measurable concentration (AUC0-last). Other pharmacokinetic parameters included AUC from time zero to infinity (AUC0-∞), time to C max (t max), apparent total body clearance of drug (CL/F), apparent volume of distribution at steady state (V ss/F), terminal rate constant (λ z ), and half-life (t 1/2). Pharmacokinetic parameters were calculated by standard non-compartmental methods of analysis using Phoenix® WinNonlin® version 5.2 (Certara Inc., Princeton, NJ, USA). Safety was assessed via evaluation of adverse events classified by Medical Dictionary for Regulatory Activities Version 13.0, physical examination, 12-lead electrocardiograms, vital signs (body temperature, supine blood pressure, and pulse rate), body weight, and standard clinical laboratory tests.
Taste was evaluated via a five-item questionnaire immediately and 10 min after the dose of atomoxetine oral solution. Each question had five possible responses. The questions assessed: (1) acceptability, or how easy the solution formulation would be to take every day, with possible responses ranging from “easy” to “not able”; (2) bitterness, with possible responses ranging from “not bitter” to “extremely bitter”; (3) sweetness, with possible responses ranging from “extremely sweet” to “not sweet”; (4) aftertaste, with possible responses ranging from “no aftertaste” to “extreme aftertaste”; and (5) flavor, with possible responses ranging from “very pleasant flavor” to “extremely unpleasant flavor.” Subjects were also able to provide additional comments on the taste of the oral solution in their own words.
2.4 Bioanalytical Methods
Plasma samples were analyzed for atomoxetine and the 4-hydroxyatomoxetine and N-desmethylatomoxetine metabolites using a liquid chromatography-tandem mass spectrometry method that was validated by inVentiv Health (Princeton, NJ, USA). In brief, the method involves solid-phase extraction of atomoxetine, 4-hydroxyatomoxetine, and N-desmethylatomoxetine from sodium-heparinized plasma. Analytes were separated by liquid chromatography using gradient elution. The liquid chromatography-tandem mass spectrometry method used the atomoxetine stable label [2H7]-LY404363 as an internal standard for atomoxetine and N-desmethylatomoxetine and used the 4-hydroxyatomoxetine stable label [2H5]-LY424478 as an internal standard for 4-hydroxyatomoxetine. The quantifiable range was 2.5–2000 ng/mL for atomoxetine and 1.0–800 ng/mL for 4-hydroxyatomoxetine and N-desmethylatomoxetine. A 10-fold dilution was also validated. For validation of the assay method, validation samples (blank plasma spiked with atomoxetine, 4-hydroxyatomoxetine, and N-desmethylatomoxetine) were analyzed across the quantifiable range to evaluate intra-/inter-run precision and intra-/inter-run accuracy. According to the clinical study protocol, plasma concentrations of N-desmethylatomoxetine and 4-hydroxyatomoxetine were measured, but statistical evaluation of bioequivalence of these metabolites was not assessed.
2.5 Statistical Analysis
All subjects who received at least one dose of study drug and had evaluable plasma concentration data were included in the pharmacokinetic analysis. In addition, analyses based on subjects who completed both treatment periods were performed. The primary pharmacokinetic parameters, Cmax and AUC0-last, and other pharmacokinetic parameters including AUC0–∞ were analyzed for the parent compound using a linear mixed-effects model, including treatment, period, and cohort as fixed effects and subject as a random effect. All pharmacokinetic data were log transformed before analysis.
The least-squares means for each formulation and the 90 % confidence interval (CI) for the difference in means between formulations were estimated from the model and back transformed from the log scale to provide estimates of the geometric means and 90 % CIs for the ratio of geometric means. Bioequivalence between the oral solution and capsule formulations was concluded if the 90 % CI of the ratio of geometric means between test and reference for both C max and AUC0-last were completely contained within the bioequivalence interval of 0.8–1.25. The statistical analysis of t max was performed using a nonparametric method (Wilcoxon signed rank test). A sample size of at least 40 subjects was selected as this was anticipated to yield more than 90 % power to show bioequivalence for both C max and AUC0-last using the 90 % CI as described.
3 Results
3.1 Subject Disposition and Demographics
A total of 42 subjects (mean age 23.2 years, range 20–37 years, body mass index 17.6–24.7 kg/m2) were randomized (21 in each cohort) and received at least one dose of study drug (Fig. 1). Two subjects enrolled in Cohort 2 received a dose of atomoxetine oral solution but did not complete the study for personal reasons. Baseline characteristics of the two cohorts were similar (Table 1).

Participant flow diagram. ATX atomoxetine
3.2 Bioequivalence
The pharmacokinetic analysis revealed that mean plasma concentration–time profiles of atomoxetine 50 mg administered as an oral solution or capsules were similar (Fig. 2). Importantly, the statistical analysis of systemic exposure for atomoxetine showed that the oral solution and capsules were bioequivalent. Specifically, 90 % CIs for the ratio of the geometric means for the primary pharmacokinetic parameters AUC0-last and C max were completely within the bioequivalence interval of 0.8–1.25 (Table 2). Further, geometric mean C max estimates were 456 ng/mL and 483 ng/mL for the oral solution and capsule formulations, respectively. In support of these findings, the statistical analysis showed that t max values for the oral solution and capsules were equal (median 0.75 h, range 0.50–6.00 h), leading to a median difference of 0 h (90 % CI 0–0.25 h; p = 0.165 between formulations). Following rapid absorption and attainment of t max, plasma concentrations of atomoxetine declined with a mean t1/2 of 2.8 h for both formulations (Table 3). Geometric mean values of other pharmacokinetic parameters including AUC0–∞, CL/F, and V ss/F were also similar for the oral solution and capsule formulations (Tables 2, 3). Similar analyses based on the 40 subjects who completed the study led to similar results (data not shown) and the same overall conclusions.

Atomoxetine plasma concentration–time profiles following administration of atomoxetine 50 mg as an oral solution or capsules. SD standard deviation
3.3 Safety
Of the 42 subjects enrolled in the study, 40 subjects completed the study and two subjects discontinued the study for personal reasons that made them unable to attend the second treatment visit. No subject discontinued the study because of an adverse event. No deaths or serious adverse events were recorded. Overall, 12 subjects reported a total of 17 adverse events considered to be related to the study drug as judged by the investigator (Table 4). All events were of mild severity and were similar in number between the oral solution and capsule formulations.
Nausea and increased blood pressure were the most common study drug-related adverse events (Table 4). No clinically significant alterations in laboratory values were noted (data not shown). The incidental findings of slightly low urine specific gravity (1.002–1.005; reference range: 1.006–1.03) in 25 subjects and slightly elevated bilirubin levels (29–32 µmol/L; reference range: 3–21 µmol/L) in seven subjects were not considered clinically significant or drug related. Regarding vital signs, mean changes in supine blood pressure were 8.7/4.6 mm Hg for the oral solution and 12.5/5.3 mm Hg for capsules at 2 h postdose. Overall, five subjects had a total of five clinically significant alterations in systolic blood pressure (SBP) ≥140 mm Hg during one of the two treatment periods. Of these, three SBP alterations occurred following administration of atomoxetine oral solution and two SBP alterations occurred with atomoxetine capsules. All events occurred within 2 h of dosing and SBP returned to normal within approximately 4 h. In addition, there was a small increase in mean supine pulse rate that was similar following treatment with both formulations (5.6 beats per minute for the oral solution and 5.5 beats per minute for capsules at 2 h postdose).
3.4 Taste Assessment
The taste (acceptability, bitterness, sweetness, aftertaste, and flavor) assessment of atomoxetine oral solution is shown in Fig. 3. Overall, acceptability of the oral solution for daily use was high. Immediately after dosing, only 3 of 42 (7.1 %) subjects indicated that the oral solution would be difficult to take every day and no subject indicated that they would not be able to take the oral solution every day. These values did not change when subjects were questioned 10 min after dosing. All subjects had at least some aftertaste both immediately after dosing and after 10 min had passed. The aftertaste was reported to be extreme or strong immediately after dosing by 26 of 42 (61.9 %) subjects and this decreased to 20 of 42 (47.6 %) subjects 10 min after dosing. According to the comments written by a few subjects, the aftertaste was bitter. Regarding flavor, a very pleasant, pleasant, or neutral flavor was reported by 34 of 42 (81.0 %) subjects immediately after dosing and by 33 of 42 (78.6 %) subjects 10 min after dosing. Only 1 of 42 (2.4 %) subjects indicated that they found the flavor to be extremely unpleasant immediately after and 10 min after dosing.

Taste and acceptability assessment of atomoxetine 50-mg oral solution
4 Discussion
This is the first reported study to confirm that a newly developed oral solution formulation of atomoxetine is bioequivalent to atomoxetine capsules. Specifically, 90 % CIs for the ratios of the primary pharmacokinetic parameters, AUC0-last and Cmax, were completely within the bioequivalence interval of 0.8–1.25 and the ratio of the geometric least-squares means were close to 1.0. Further, the median and range values of t max for atomoxetine oral solution and capsules were equal and geometric mean values of other pharmacokinetic parameters were also similar for oral solution and capsule formulations. Furthermore, results of the safety analysis did not raise any new safety concerns with atomoxetine oral solution. Finally, an exploratory analysis found that the taste acceptability of the oral solution was high despite the existence of aftertaste effects noted by a high proportion of study subjects.
A diagnosis of ADHD in children commonly occurs at around 6–7 years of age. At this age, children often have difficulty swallowing solid formulations such as capsules. In support of this, a medication acceptance survey indicated that the percentage of children/adolescents who were able to easily swallow solid formulations, particularly capsules, decreased with a younger age and was especially notable in children younger than 9 years of age [18]. Further, there is good evidence that solid formulations are less acceptable than liquid formulations in children [15, 22]. Not surprisingly, a survey among Japanese ADHD specialists regarding the medical needs for atomoxetine in Japan found that “development of a formulation that is easy to swallow” was ranked as the second highest need [17]. In a study of acceptability of medicines in the pediatric patients, 31 % of parents and caregivers reported refusal on at least one occasion [23]. Therefore, providing a formulation that is easier for children to take is very important for starting and maintaining ADHD treatment. Further, there is a requirement for pediatric assessments during medication development to use “appropriate formulations for each age group for which the assessment is required” [14], and that young patients “should be treated with medicinal products of which the pharmaceutical design is tailored for their age group” [13]. Based on the need for an additional formulation suitable for younger children with ADHD, an oral solution of atomoxetine was developed. The results of this study, demonstrating bioequivalence of the oral solution and capsule formulations of atomoxetine, are a critical step towards making this formulation available for patients that can benefit from it and addresses this clinical need.
In addition to helping overcome problems with swallowing capsules, an oral solution may also help ensure children receive individualized doses based on body weight. Atomoxetine capsules are available in a wide range of marketed strengths (5, 10, 18, 25, 40, 60, 80, and 100 mg) to help facilitate accurate dosing, especially for children who weigh less than 70 kg. Dosing with atomoxetine oral solution will allow for even more accurate dosing for such children.
In this study, a taste assessment was included to collect general taste acceptance data and because there was a concern that the raspberry flavor of the oral solution used for taste masking is not familiar to most Japanese people and, therefore, may not be acceptable. The results of the taste assessment suggest that the palatability of atomoxetine oral solution is generally favorable, with no subjects responding that they would not be able to take this medicine every day. Hence, the development of atomoxetine oral solution has addressed one of the key challenges that needs to be overcome when developing liquid formulations [16, 19]. Palatability is critical to acceptability and is mainly determined by taste [15]. Taste is an important aspect to address when developing liquid formulations because “unpleasant taste of medication” has been cited as a key barrier that affects adherence by 83.9 % of surveyed pediatricians [24]. Poor palatability, as one cause of repetitive resistance among children, has also been cited as both a barrier to adherence and a source of stress/caregiver fatigue by caregivers of children with chronic illnesses [25].
This study employed a cross-over design, which is the standard design used to assess the bioequivalence between two formulations. The dose selected in this study (50 mg) was selected to resemble the typical dose delivered twice daily that would be suitable for Japanese adults. In this study, CYP2D6 extensive metabolizers, which included intermediate metabolizers and ultrarapid metabolizers, were recruited and poor metabolizers were excluded. The prevalence of poor metabolizers is very low in Japan, so this covers the majority of the Japanese population. Furthermore, the CYP2D6 polymorphism has been shown to influence the pharmacokinetics of atomoxetine, and poor metabolizers have a lower clearance; therefore, this inclusion restriction is in accordance with advice that subjects with higher clearance should be evaluated if drug clearance differs to a large extent among subjects owing to genetic polymorphism [20]. Atomoxetine can be dosed to provide similar efficacy and safety in extensive and poor metabolizer patients without knowing their metabolizer status [26].
In this study, adults but not children were studied. When comparing formulations, studies are usually conducted in adults. A previous study has shown that atomoxetine pharmacokinetics in pediatric and adult extensive metabolizer patients were similar after adjustment for body weight [27]. Although taste acceptability was important in this study, children are known to have different taste preferences from adults, including a greater preference for sweet tastes and rejection of bitter tastes [28]. Therefore, the results from the taste assessment in this study need to be verified in the population that will actually use the oral solution formulation. However, these data at least serve as a rough guide regarding the overall acceptance of the formulation and the perception of raspberry taste in Japanese subjects.
Regarding safety, these results do need to be extrapolated to female patients and patients with different metabolizer genotypes because the population in this bioequivalence study excluded poor metabolizer and was limited to male subjects. However, atomoxetine can be dosed to provide similar efficacy and safety levels in extensive and poor metabolizer patients without knowledge of metabolizer status [26], the dose in children are adjusted for body weight, and there is no dose adjustment by sex [26].
5 Conclusion
This study showed that atomoxetine oral solution was bioequivalent to atomoxetine capsules. In addition, the two formulations were similarly well tolerated and atomoxetine oral solution was found to be generally palatable, which addresses one of the challenges of developing liquid formulations. In clinical practice, atomoxetine oral solution may be an alternative formulation for children and any individuals with ADHD who have actual or anticipated difficulties in swallowing atomoxetine capsules.
References
Adler LA, Spencer TJ, Williams DW, Moore RJ, Michelson D. Long-term, open-label safety and efficacy of atomoxetine in adults with ADHD: final report of a 4-year study. J Atten Disord. 2008;12:248–53.
Bangs ME, Emslie GJ, Spencer TJ, Ramsey JL, Carlson C, Bartky EJ, Busner J, Duesenberg DA, Harshawat P, Kaplan SL, Quintana H, Allen AJ, Sumner CR. Efficacy and safety of atomoxetine in adolescents with attention-deficit/hyperactivity disorder and major depression. J Child Adolesc Psychopharmacol. 2007;17:407–20.
Kratochvil CJ, Vaughan BS, Stoner JA, Daughton JM, Lubberstedt BD, Murray DW, Chrisman AK, Faircloth MA, Itchon-Ramos NB, Kollins SH, Maayan LA, Greenhill LL, Kotler LA, Fried J, March JS. A double-blind, placebo-controlled study of atomoxetine in young children with ADHD. Pediatrics. 2011;127:e862–8.
Montoya A, Hervas A, Cardo E, Artigas J, Mardomingo MJ, Alda JA, Gastaminza X, Garcia-Polavieja MJ, Gilaberte I, Escobar R. Evaluation of atomoxetine for first-line treatment of newly diagnosed, treatment-naive children and adolescents with attention deficit/hyperactivity disorder. Curr Med Res Opin. 2009;25:2745–54.
Prasad S, Harpin V, Poole L, Zeitlin H, Jamdar S, Puvanendran K, Group SS. A multi-centre, randomised, open-label study of atomoxetine compared with standard current therapy in UK children and adolescents with attention-deficit/hyperactivity disorder (ADHD). Curr Med Res Opin. 2007;23:379–94.
Takahashi M, Goto T, Takita Y, Chung SK, Wang Y, Gau SS. Open-label, dose-titration tolerability study of atomoxetine hydrochloride in Korean, Chinese, and Taiwanese adults with attention-deficit/hyperactivity disorder. Asia Pac Psychiatry. 2014;6:62–70.
Goto T, Hirata Y, Takita Y, Trzepacz PT, Allen AJ, Song DH, Gau SS, Ichikawa H, Takahashi M. Efficacy and safety of atomoxetine hydrochloride in Asian adults with ADHD: a multinational 10-week randomized double-blind placebo-controlled Asian study. J Atten Disord. 2013;. doi:10.1177/1087054713510352 (Epub ahead of print).
Michelson D, Adler L, Spencer T, Reimherr FW, West SA, Allen AJ, Kelsey D, Wernicke J, Dietrich A, Milton D. Atomoxetine in adults with ADHD: two randomized, placebo-controlled studies. Biol Psychiatry. 2003;53:112–20.
Buitelaar JK, Wilens TE, Zhang S, Ning Y, Feldman PD. Comparison of symptomatic versus functional changes in children and adolescents with ADHD during randomized, double-blind treatment with psychostimulants, atomoxetine, or placebo. J Child Psychol Psychiatry. 2009;50:335–42.
Gau SS, Shang CY. Improvement of executive functions in boys with attention deficit hyperactivity disorder: an open-label follow-up study with once-daily atomoxetine. Int J Neuropsychopharmacol. 2010;13:243–56.
Maziade M, Rouleau N, Lee B, Rogers A, Davis L, Dickson R. Atomoxetine and neuropsychological function in children with attention-deficit/hyperactivity disorder: results of a pilot study. J Child Adolesc Psychopharmacol. 2009;19:709–18.
Svanborg P, Thernlund G, Gustafsson PA, Hagglof B, Schacht A, Kadesjo B. Atomoxetine improves patient and family coping in attention deficit/hyperactivity disorder: a randomized, double-blind, placebo-controlled study in Swedish children and adolescents. Eur Child Adolesc Psychiatry. 2009;18:725–35.
European Medicines Agency. Guideline on pharmaceutical development of medicines for paediatric use. 11 May 2011. EMA/CHMP/QWP/180157/2011. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/06/WC500107908.pdf. Accessed 2 Feb 2015.
Pediatric Research Equality Act. One Hundred Tenth Congress of the United States of America. http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DevelopmentResources/UCM049870.pdf. Accessed 5 Feb 2015.
Ernest TB, Elder DP, Martini LG, Roberts M, Ford JL. Developing paediatric medicines: identifying the needs and recognizing the challenges. J Pharm Pharmacol. 2007;59:1043–55.
Nunn T, Williams J. Formulation of medicines for children. Br J Clin Pharmacol. 2005;59:674–6.
Miyachi T, Miyajima T, Ishizaki Y, Fukai Y, Ishiwaki A, Nagai A, Tanaka H. The investigation into the actual conditions of medication for the child with attention deficit/hyperactivity disorder (AD/HD) in Japan [in Japanese]. Psychiatria et Neurologia Paediatrica Japonica. 2010;50:419–27.
Polaha J, Dalton WT 3rd, Lancaster BM. Parental report of medication acceptance among youth: implications for everyday practice. South Med J. 2008;101:1106–12.
Walsh J, Cram A, Woertz K, Breitkreutz J, Winzenburg G, Turner R, Tuleu C, European Formulation Initiative. Playing hide and seek with poorly tasting paediatric medicines: do not forget the excipients. Adv Drug Deliv Rev. 2014;73:14–33.
Ministry of Health, Labour and Welfare. PFSB/ELD Notification No. 1124004, Guideline for Bioequivalence Studies of Generic Products, November 24, 2006. http://www.nihs.go.jp/drug/be-guide(e)/be2006e.pdf. Accessed 3 Jul 2016.
Pharmaceutical and Food Safety Bureau. Guideline for bioequivalence studies of generic products. Available from: http://www.nihs.go.jp/drug/be-guide(e)/Generic/GL-E_120229_BE_rev140409.pdf. Accessed 5 Feb 2015.
Beck MH, Cataldo M, Slifer KJ, Pulbrook V, Guhman JK. Teaching children with attention deficit hyperactivity disorder (ADHD) and autistic disorder (AD) how to swallow pills. Clin Pediatr. 2005;44:515–26.
Venables R, Batchelor H, Hodson J, Stirling H, Marriott J. Determination of formulation factors that affect oral medicines acceptability in a domiciliary paediatric population. Int J Pharm. 2015;480:55–62.
American Academy of Pediatrics. Periodic Survey #44: patient compliance with prescription regimens. Available from: http://www.aap.org/en-us/professional-resources/Research/Pages/PS44_Executive_Summary_PatientCompliancewithPrescriptionRegimens.aspx. Accessed 3 Feb 2015.
Santer M, Ring N, Yardley L, Geraghty AW, Wyke S. Treatment non-adherence in pediatric long-term medical conditions: systematic review and synthesis of qualitative studies of caregivers’ views. BMC Pediatr. 2014;14:63.
Trzepacz PT, Williams DW, Feldman PD, Wrishko RE, Witcher JW, Buitelaar JK. CYP2D6 metabolizer status and atomoxetine dosing in children and adolescents with ADHD. Eur Neuropsychopharmacol. 2008;18:79–86.
Witcher JW, Long A, Smith B, Sauer JM, Heilgenstein J, Wilens T, Spencer T, Biederman J. Atomoxetine pharmacokinetics in children and adolescents with attention deficit hyperactivity disorder. J Child Adolesc Psychopharmacol. 2003;13:53–63.
Mennella JA, Pepino MY, Reed DR. Genetic and environmental determinants of bitter perception and sweet preferences. Pediatrics. 2005;115:e216–22.
Acknowledgments
The authors wish to acknowledge Amanda Long for her support of the pharmacokinetic analysis and the clinical site investigator. Medical writing assistance was provided by Mark Snape, MBBS, CMPP, and Justine Southby, PhD, CMPP, of ProScribe-Envision Pharma Group and was funded by Eli Lilly. ProScribe’s services complied with international guidelines for Good Publication Practice (GPP2).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was sponsored by Eli Lilly Japan K.K., manufacturer/licensee of Strattera® (atomoxetine hydrochloride).
Conflicts of interest
All authors are employees of either Eli Lilly Japan K.K. (MN, YS, TG) or Eli Lilly and Company (JW) and declare ownership of stock or equity in Eli Lilly and Company, Indianapolis, IN, USA.
Ethical Approval
All procedures performed were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Approval of the study by the appropriate ethical review boards was granted.
Informed Consent
Written informed consent was obtained from all individual participants included in the study.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Nakano, M., Witcher, J., Satoi, Y. et al. Pharmacokinetic Profile and Palatability of Atomoxetine Oral Solution in Healthy Japanese Male Adults. Clin Drug Investig 36, 903–911 (2016). https://doi.org/10.1007/s40261-016-0430-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-016-0430-y