
Abstract
Copy number variants (CNVs) have been identified as a major risk factor in neuropsychiatric disorders. In this review, we describe the phenotypes and syndromic features associated with CNVs at four of the best-characterized risk loci for these disorders: 15q11.2-13.1, 22q11.2, 16p11.2, and 7q11.23. By considering the reported prevalence of these CNVs in autism, intellectual disability, schizophrenia, and controls, we demonstrate a pattern of asymmetric shared risk in which CNVs increase the risk of multiple disorders but to differing degrees. This asymmetric risk sharing is incompatible with a model in which CNVs observed in autism or schizophrenia are secondary to a reduction in IQ, but favors a more complex relationship between individual CNVs and specific neuropsychiatric phenotypes. Finally, we discuss how the lessons learned from CNVs in neuropsychiatric disorders will translate to the expanding list of genes being associated with these disorders through exome sequencing.
Similar content being viewed by others


Introduction
Neuropsychiatric conditions such as intellectual disability (ID), autism spectrum disorder (ASD), and schizophrenia are among the most common and debilitating of human disorders, yet they are also among the least well understood. All three conditions are known to have a strong genetic component [1]; and with recent advances in technology, the genetic risk factors underlying this heritability are gradually being identified. Despite the diagnostic differences, many of the genetic risk factors are shared among these disorders.
In this review, we will consider the phenotypic overlap associated with four of the best-characterized neuropsychiatric genetic risk factors, specifically copy number variants (CNVs) at 22q11.2, 15q11.2-13.1 (BP2-3), 16p11.2 (BP4-5), and 7q11.23 (Table 1). We will consider how understanding this overlap can inform our understanding of the architecture of risk in these conditions, and the implications as specific genes are implicated with each disorder.
Chromosomal Abnormalities and Copy Number Variants (CNVs)
Historically syndromes caused by chromosomal abnormalities, identified through karyotype analysis, have been associated with several neuropsychiatric conditions. For example, Down syndrome, caused by an extra copy of chromosome 21 [2] is a common cause of ID and, less commonly, of ASD [3].
Alongside detecting abnormal numbers of chromosomes, karyotype analysis is also used to identify chromosomal rearrangements, such as translocations, where large segments of DNA are swapped between chromosomes or within the same chromosome. Such translocations were used to demonstrate that a region on the long arm of chromosome 22 [4, 5] was involved in DiGeorge syndrome, a condition that is frequently associated with ID [6] alongside numerous other syndromic features.
As technology has progressed, it has become possible to identify smaller changes in chromosomal structure such as deletions and duplications, collectively called copy number variations (CNVs). High-resolution karyotyping allows genome-wide detection of large CNVs (e.g., >2–5 Mb), while fluorescence in situ hybridization (FISH) allows targeted detection of sub-microscopic CNVs (e.g., >100 kb). Many such CNVs are clinically relevant, for example, deletion CNVs on the long arm of chromosome 22 (22q11.2) are a more common cause of DiGeorge syndrome than the translocations identified by karyotype analysis [7].
The advent of chromosomal microarrays has allowed reliable detection of smaller CNVs (e.g., >30 kb) in a genome-wide manner, i.e., without having to select a specific target region as for FISH. Application of this technique to large cohorts has demonstrated that large CNVs are associated with ID [8•], ASD [9, 10••, 11], and schizophrenia [12, 13] and that many of these CNVs are de novo mutations observed in the affected child and not in either parent. Furthermore, by identifying CNVs that are recurrent in multiple affected individuals new loci have been associated with neuropsychiatric disorders, including 1q21.1 [14], 16p11.2 [15–18], 7q11.23 duplications [10••], and 17q12 deletions [19]. These recurrent loci are often caused by a process called non-allelic homologous recombination (NAHR) in which segmental duplications (large regions of very similar DNA) in close proximity lead to DNA replication errors resulting in deletion or duplication of the intervening unique DNA [20].
We will focus on deletion and duplication CNVs at four loci, 22q11.2, 15q11.2-13.1 (BP2-3), 16p11.2 (BP4-5), and 7q11.23 (Table 1), that show clear association with neuropsychiatric conditions and occur frequently enough to make a comparison between ID, ASD, schizophrenia, and unaffected controls.
Prevalence of CNVs by Disorder
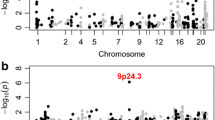
To date these four CNVs have not been systematically assessed in a large birth cohort to determine the true population incidence and phenotypic spectrum. The prevalence in unaffected individuals has been estimated in a population of 101,655 individuals [21••], while the prevalence in individuals with neuropsychiatric disorders has been estimated in 3,955 individuals with idiopathic ASD [22], and 4,719 individuals with schizophrenia [23•]. No comparable cohort of ID cases has been assessed, therefore, we will rely on 8,772 individuals referred for microarray testing, predominantly due to ID with or without syndromic features [8•]. Due to the manner by which this ID cohort was ascertained the rate of CNVs is likely to be inflated, however, at present, it is the best dataset available. The observed prevalence of each deletion and duplication CNV at each of the four loci is shown for ID, ASD, schizophrenia, and controls in Fig. 1a; the corresponding odds ratios are shown in Table 2.

Prevalence of CNVs in three neuropsychiatric disorders and controls. a The prevalence of eight CNVs at four loci is shown for individuals with schizophrenia (Sch, n = 4,719), intellectual disability (ID, n = 8,772), autism spectrum disorder (ASD, n = 3,955), and in controls (n = 101,655). Some CNVs increase the risk of all three disorders (e.g., 16p11.2 duplications, 7q11.23 duplications) others show a degree of specificity being rarely observed in schizophrenia (16p11.2 deletions, 22q11.2 duplications, 15q11.2-13.1 duplications), or rarely observed in ASD and schizophrenia (7q11.23 deletions). b The expected pattern of CNV prevalence based on four models of etiology described in the main text. The "trunk and branches" model shows a pattern of asymmetric sharing that matches many of the CNVs in (a)
22q11.2: Three Syndromes Unified by a Genetic Diagnosis
Deletions at 22q11.2 are observed in about 1 in 6,000 births [24] and result in a wide variety of phenotypes, so much so that three separate syndromes were described. DiGeorge syndrome was described in 1967 based on a severe immune deficiency and absent thymus; hypocalcemia, hypoparathyroidism, and a right aortic arch were recorded in some infants. The authors noted that abnormal development of the third and fourth pharyngeal pouches could explain most of these features [25]. A decade later abnormalities of the heart and a distinctive facial appearance were described as cono-truncal anomaly face syndrome (CTAF) in Japan [26], while 2 years later velo-cardio-facial syndrome (VCFS) was described based on abnormalities of the palate (velum), heart, and face [27], it was also noted that the most of the children had learning disabilities. As mentioned earlier, the detection of a translocation that involved chromosome 22 [4] followed by deletions detected using high-resolution karyotyping and FISH allowed DiGeorge syndrome [28, 29], CTAF [30], and VCFS [31] to be recognized as sharing a common genetic cause: deletions at 22q11.2.
The deletion also leads to a wide spectrum of neurodevelopmental phenotypes, including ID, ASD, and schizophrenia [32] (Fig. 1a); of note, a lower IQ score appears to be associated with the increased risk of schizophrenia-spectrum psychosis [32].
Duplications at 22q11.2 are considerably more common being observed in about 1 in 1,300 adults [21••]. The phenotype is highly variable, including ID, speech impairment, psychomotor delay, growth retardation, hypotonia, and non-specific dysmorphic features [33]. It is frequently observed in individuals without neuropsychiatric diagnoses and is the least penetrant of the eight CNVs discussed in this review [21••]. It has recently been noted that the duplication was observed at a lower frequency in schizophrenia (0.014 %, 3 in 21,138 cases) than in unaffected controls (0.085 %, 23 in 27,133 controls) raising the possibility of imparting protection. An alternative interpretation is that, in the presence of other neuropsychiatric risk factors, 22q11.2 duplications tip the balance towards ID and ASD rather than schizophrenia.
15q11.2-13.1: Syndromes that Vary with Imprinting
Multiple segmental duplications are observed in the 15q11.2-13.1 region leading to five distinct "breakpoints" capable of generating recurrent large CNVs. We will focus only on the largest CNVs that include the region between breakpoint 2 and 3 (Table 1). In addition, the region between these two breakpoints is imprinted, which means that some of the genes within it are only expressed on the maternal or paternal chromosomes. Accordingly deletions give rise to two phenotypically distinct syndromes, Angelman and Prader–Willi, depending which parent’s chromosome the deletion is on.
A deletion on the maternal chromosome results in Angelman syndrome (AS), which is diagnosed in 1 in 10,000 to 1 in 20,000 live births [34, 35]. This syndrome was first described in 1965 [36] by the combination of distinctive behaviors and symptoms in three children. The disorder is characterized by severe ID with limited or absent speech, gait ataxia, seizures with a characteristic EEG pattern consisting of high voltage slow-wave activity, microcephaly, and hypopigmentation. Additionally, people with this condition exhibit an apparently happy disposition with excitability and inappropriate laughter; ASD is present in over 50 % of cases [37]. The association with 15q11.2-13.1 was detected through high-resolution karyotype analysis [38]. Analysis of an AS patient with a 15q inversion [39] and candidate gene mutation screening [40] determined that disruption of the ubiquitin ligase gene UBE3A within the 15q11.2-13.1 region was sufficient to cause AS. Only the maternal copy of UBE3A is expressed in the brain [41], though in the lymphoblasts and fibroblasts both copies are expressed.
Conversely, a deletion on the paternal chromosome leads to Prader–Willi syndrome (PWS), which is diagnosed in about 1 in 20,000 live births [42]. PWS was clinically described in nine patients with distinctive symptoms in 1965 [43]. It is characterized by neonatal hypotonia, low birth weight, and failure to thrive as an infant, mild ID, small hands and feet, hypopigmentation, and hypogonadism. As the individual enters early childhood, the failure to thrive is replaced by hyperphagia (excessive eating) that results in severe obesity [44]. In addition, these individuals have a distinct behavioral phenotype characterized by repetitive and stereotyped behaviors; up to 35 % of them receive a diagnosis of ASD [45]. The association with 15q11.2-13.1 was identified through high-resolution karyotype analysis [46] and the RNA processing gene SNURF-SNRPN or proximal small nucleolar RNAs (snoRNAs) are implicated in many symptoms [47].
Both AS and PWS can also arise through other processes including: (1) Uniparental disomy of chromosome 15, in which two chromosomes are present (as normal), however, they both come from the same parent; (2) Small mutations that disrupt the imprinting center on 15q11.2; and (3) Small mutations in UBE3A on the maternal chromosome leading to AS [48, 49] or in SNURF-SNRPN or snoRNAs on the paternal chromosome possibly leading to PWS [47, 49]. Of note, no cases of AS or PWS were identified in the ASD cohort (Fig. 1a); this is likely a consequence of the comparative rarity of these CNVs compared with other loci.
Duplications of 15q11.2-13.1 are observed in ~1 in 8,000 adults [21••]. These duplications were found in several cases of ASD [50] by karyotype analysis, but also in unaffected parents [51] raising doubt about the ASD association. The observation that duplications on the maternal chromosome carry considerably greater risk than those on the paternal chromosome [52] resolved this lack of segregation; maternal duplications are now recognized as one of the most frequent risk factors for ASD (Fig. 1a). While the features are less specific than for AS or PWS, a clear syndrome is characterized by ASD, ID, abnormal speech development, behavioral disturbances, seizures, hypotonia, short stature, and attention deficit and hyperactivity disorder (ADHD). Concerningly, several cases of sudden death have been reported in individuals with 15q11.2-13.1 duplications [53] though the etiology of this phenomenon remains unknown. The duplication can occur by two mechanisms: (1) An additional supernumerary marker chromosome, also known as isodicentric chromosome 15 (IDIC15), with two extra copies leading to a total of four copies of the 15q11.2-13.1 region; and (2) An interstitial duplication leading to a total of three copies of the region.
16p11.2: A Syndrome Discovered by Genomics
CNVs at 16p11.2 are the most common of these four loci (Fig. 1a), however, the association with neuropsychiatric disorders was only demonstrated in 2008 [15–17, 54]. The deletion is observed in 1 in 3,500 live births [55] and was initially described as a cause of idiopathic ASD. Subsequently, it has been identified in ID [56], schizophrenia [18], and a host of other neuropsychiatric conditions. As more individuals have been examined a syndrome has emerged including obesity, ID, speech problems, motor delays, and macrocephaly along with a variety of congenital abnormalities [57•]. The deletion is also observed in individuals without ID, ASD, or schizophrenia [21••]; this was initially described as incomplete penetrance, however, careful examination of carriers compared with non-carrier family controls showed a consistent decrease in IQ (by about two standard deviations) and social impairment (1.6 standard deviations on the SRS) [57•]. If all developmental and neuropsychiatric conditions are included in the definition of "affected" then 16p11.2 deletions are over 95 % penetrant [57•].
The 16p11.2 duplication is observed in 1 in 1,350 live births [55]. Like the 16p11.2 deletions, they were initially detected in individuals with idiopathic ASD [15, 17], but subsequently this CNV has been associated with many neurodevelopmental disorders including ID, speech delay, epilepsy, bipolar disorder, schizophrenia, ADHD, anxiety disorder, motor delay, and spastic quadriparesis [18, 56, 58]. The phenotype also includes low BMI, microcephaly, failure to thrive, congenital anomalies, and mild dysmorphic features [56].
While both the deletion and duplication increase risk for neuropsychiatric disorders, in particular ID, they display mirror phenotypes for head circumference and body mass index (BMI) with the combination of high BMI and macrocephaly in the deletion, and low BMI and microcephaly in the duplication [56, 59]. These relationships raise the possibility that these phenotypes are directly correlated with 16p11.2 gene dosage and possibly expression of specific genes.
7q11.23: A Genetic Locus for Human Sociability
Deletions at 7q11.23 cause Williams–Beuren syndrome (WBS) which was described in the early 1960′s [60, 61] and is characterized by supravalvular aortic stenosis, ID (with verbal IQ comparatively spared), hypersociable personality, growth retardation, endocrine abnormalities, and a distinctive facial appearance [62]. The causal role of 7q11.23 deletions was discovered through FISH analysis of the Elastin (ELN) gene in WBS patients following the recognition of ELN disruption as a cause of familial supravalvular aortic stenosis [63]. The disorder affects about 1 in 10,000 individuals [62]. Individuals with WBS can have a concurrent diagnosis of ASD [64], however, this is uncommon [62].
While the phenotype of deletions at 7q11.23 was recognized as a distinct syndrome over 50 years ago, the first description of a phenotype associated with duplications at this locus was in 2005 [65]. Analysis of multiple cases has revealed a new syndrome characterized by severe speech delay, mild ID, and non-specific dysmorphic features. Of note, the individuals tended to exhibit poor eye contact and to be withdrawn [66, 67]. The population frequency is estimated to be similar to that for WBS, affecting 1 in 13,000–1 in 20,000 individuals [67]. Duplications at 7q11.23 have been associated with ASD [19] and schizophrenia [68].
In a similar manner to 16p11.2 CNVs, both the deletion and duplication CNVs at 7q11.23 have an adverse effect on an individual’s IQ, but a mirror phenotype is seen with sociability and speech. Sociability is increased and verbal IQ is relatively preserved in the deletion, while sociability is reduced and there is severe speech impairment in the duplication. Again this raises the possibility of direct correlation between these phenotypes and gene dosage/expression.
Multiple Risk Factors
These four CNV loci are among the most common risk factors for neuropsychiatric disorders, however, together they account for <2.5 % of patients with these disorders. In ASD alone it is estimated that up to 1,000 distinct genetic risk factors may contribute to the phenotype [69, 70], and there is no reason to expect radically different figures in ID or schizophrenia.
Despite being treated as single diagnostic entities, each affected individual is likely to have a unique combination of risk factors. In ASD, this etiological complexity has led to the term “the autisms” [71] to reflect this extensive phenotypic and genetic heterogeneity contained in the single diagnostic label.
However, aspects of the phenotype are sufficiently similar to allow the use of ID, ASD, and schizophrenia as diagnoses, therefore, the myriad risk factors must converge on a limited number of pathophysiological mechanisms that cause the core features of each condition. Several groups have attempted to leverage this convergence by seeking stages of brain development and anatomical regions of the brain at which known genetic risk factors are expressed. Such convergence has been observed in the pre-frontal cortex during mid-fetal development in both ASD [72–74] and schizophrenia [75]. These approaches show the potential for rigorously identified genetic risk factors to be leveraged to reveal fundamental insights into the pathology of neuropsychiatric disorders.
Penetrance in Neuropsychiatric Traits
These syndromes challenge the conventional notion of penetrance. While 16p11.2 deletions show incomplete penetrance for ID and ASD (when viewed as dichotomous, qualitative traits) they appear to cause a significant deleterious impact in cognition and social behavior, as measured by IQ and the Social Responsiveness Scale (SRS) (both of which are continuous, quantitative traits), in both cases and apparently unaffected carriers [57•]. While the disorder is incompletely penetrant, the effect is completely penetrant. Similarly when unaffected carriers discovered in a population-scale search for CNVs were examined, they displayed a higher level of cognitive deficits than individuals without such CNVs [21••]. This concept of penetrance across multiple neurodevelopmental and neuropsychiatric disorders and the use of continuous, quantitative measures such as SRS and IQ to compare probands with their unaffected first-degree relatives is described further in the Developmental Brain Dysfunction model [76••].
Phenotypic Overlap
One of the most unexpected and striking findings from analysis of CNVs in neuropsychiatric disorders has been the extent to which specific loci are shared between disorders. For example, deletions at both 22q11.2 (DiGeorge syndrome) and 16p11.2 increase the individual’s risk for ID [6, 77], ASD [10••, 78••], and schizophrenia [6, 23•]. Furthermore, while the presence of these CNVs in ASD cohorts does not correlate with ASD severity, they do correlate (weakly) with IQ [10••]. These observations challenge the notion that ID, ASD, and schizophrenia are three entirely distinct diagnostic and etiological categories. We will consider whether the CNV data support or contradict four models for the relationship between neuropsychiatric phenotype and etiological mechanism.
Diagnostic Unity
The simplest explanation is that these three neuropsychiatric disorders are, in fact, a single etiological entity. Clinical observations would argue against this; a diagnosis of ASD is rarely converted to a diagnosis of schizophrenia [79], which holds true in a reciprocal fashion. Similarly individuals with ASD or schizophrenia can have a high IQ. The genetic evidence is also inconsistent with this explanation. We would expect each CNV to be equally prevalent in all three conditions (Fig. 1b), however, with the exception of 16p11.2 duplications, this does not appear to be the case (Fig. 1a).
IQ Buffering in ASD and Schizophrenia
Given the consistency with which de novo mutations are associated with ID (Fig. 1a) and the correlation of de novo mutations with IQ in ASD [10••] and, possibly, schizophrenia [80] it could be argued that the primary effect of de novo CNVs is to reduce IQ leading to ID directly and ASD and schizophrenia indirectly. In this model [81] limited social skills or the tendency to disordered thought patterns are distributed normally within the population, but a higher IQ has a buffering effect that masks the phenotype. A de novo CNV disrupts this buffering effect potentially leading to ASD or schizophrenia. Under this model, we would expect each de novo CNV to be most prevalent in ID and for ASD and schizophrenia to mirror this risk (Fig. 1b) however, no CNVs show this pattern clearly.
IQ Buffering in ASD only
This is the same concept as the last model, however, given the closer relationship between IQ and ASD than between IQ and schizophrenia we will consider the IQ buffering effect to only relate to ASD. At least two CNVs appear to show a consistent pattern (16p11.2 deletions, 7q11.23 duplications), however, the majority of CNVs show a more complex pattern (Fig. 1a).
Trunk and Branches
De novo CNVs can disrupt key processes in brain development (the trunk), thus raising the risk for multiple neuropsychiatric disorders [76••], but they can also disrupt more specific neurological processes (the branches), e.g., social skills, that further increase the risk for specific subsets of neuropsychiatric disorders. Consequently, de novo CNVs show a pattern of asymmetric sharing in which they are observed in multiple neuropsychiatric disorders, but are particularly prevalent in a subset of disorders. Most CNVs show this asymmetric sharing: 15q11.2-13.1 duplications disproportionately increase the risk of ASD, 22q11.2 deletions increase the schizophrenia risk more than ASD risk, and 22q11.2 duplications predominantly increase ID risk and may even be protective against schizophrenia [82].
Therefore, while some CNVs increase the general risk of neuropsychiatric disorders, the majority appear to increase the risk of certain conditions more than others, suggesting that at least some genetic risk factors may be specific to each disorder. These CNV risk factors not only modify an individual’s position on continuous neuropsychiatric traits but also do so in an asymmetric manner, affecting some traits to a greater extent than others. The extent of this sharing between phenotypes may reflect the degree to which etiological mechanisms are shared between disorders.
"Missed Syndromes"
While some of these CNVs cause well-recognized syndromes (DiGeorge, WBS, AS, PWS) others, such as 16p11.2 and 7q11.23 duplications, were discovered in cases of idiopathic ASD or schizophrenia without apparently syndromic features. It was only with the examination of large cohorts of individuals that share the same CNV that a syndrome has emerged.
This raises a question regarding which factors separate recognizable syndromes from those that are "missed." The clearest factor would appear to be the presence of distinctive, uncommon, and specific characteristics. For example, WBS accounts for a substantial proportion of individuals with supravalvular aortic stenosis and is associated with a distinctive appearance and behaviors. Similarly the distinctive combination of absent thymus and hypoparathyroidism allowed the recognition of DiGeorge syndrome [25]. Conversely, the abnormalities in cognition, behavior, and energy balance observed in CNVs at 16p11.2 are observed relatively frequently in the population, such that large cohorts of cases are required to demonstrate syndromic features [57•, 59].
Population frequency of the syndrome must play a role, since several cases must be brought to the attention of the clinician and/or researcher, however, above this threshold population frequency would appear to be less important than distinctive features, since 16p11.2 is three times more common than 15q11.2-13.1 but was recognized considerably later. Penetrance might also be expected to play a role, since a highly penetrant variant will show a clear segregation with the disorder allowing easier recognition. While the late discovery of syndromes for 16p11.2 and 22q11.2 duplications would support this, 7q11.23 duplications have the highest penetrance of all eight CNVs (Table 1), but were discovered almost last.
In summary, a syndrome has emerged for CNVs at all four loci associated with neuropsychiatric disorders after sufficient cases have been examined, however, only syndromes with particularly distinctive features are identified prior to genomic analysis of large cohorts.
Single Gene Mutations
Exome sequencing in ID [83, 84], ASD [69, 85–87], and schizophrenia [80] has highlighted the role of de novo loss of function mutations as risk factors for these disorders. Furthermore, by observing multiple mutations in the same gene in different individuals, specific genes have been associated with disorders [69].
As with de novo CNVs, no single gene will explain a large proportion of ID, ASD, or schizophrenia; FMR1 triplet expansions (Fragile X) are the most common single gene cause of ID [88] and ASD [89, 90] accounting for only 4 % and 1 % of cases, respectively. Unlike the CNVs, most of the identified genes have no equivalent mutations in controls suggesting 100 % penetrance, however, with increasing sample size this is likely to change.
It is less clear whether single gene mutations will show similar patterns of asymmetric sharing between disorders as is observed with CNVs (Fig. 1a). This mixture of phenotypic effects may be a consequence of the multiple genes within a CNV; if this were the case, we might expect to see a group of genes that increase the general risk of neuropsychiatric disorders (disrupting the "trunk") and another group that are specific to one disorder (disrupting the "branches"). Alternatively the pleiotropic effects of each gene might lead to similarly diverse patterns of prevalence within these disorders as is seen with CNVs.
Finally, it is unclear whether careful examination of individuals with disruption of the same gene will lead to a recognizable syndrome or not. Some of the syndromic features appear to be a consequence of the multiple genes within each CNV. However, disruption of a single gene can certainly cause syndromic effects (e.g., FMR1, TSC1); it remains to be seen if these genes represent the exception or the rule.
Conclusion
Advances in technology have allowed systematic identification of CNVs that mediate risk for neuropsychiatric disorders leading to the recognition of novel syndromes (16p11.2, 7q11.23 duplications). The identification of these CNVs has transformed our understanding of the genomic architecture of these disorders leading to the conclusion that thousands of risk factors converge on a limited number of diagnoses. Furthermore, a complex pattern of asymmetric sharing of genetic risk factors between neuropsychiatric disorders suggests that ID, ASD, and schizophrenia are caused by a combination of shared and distinct neuropathological mechanisms.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Carroll LS, Owen MJ. Genetic overlap between autism, schizophrenia and bipolar disorder. Genome Med. 2009;1:102.
Lejeune J, Gauthier M, Turpin R. Les chromosomes humains en culture de tissus, vol. 248. Paris: Gauthier-Villars/Editions Elsevier; 1959. p. 602–3.
Lowenthal R, Paula CS, Schwartzman JS, Brunoni D, Mercadante MT. Prevalence of pervasive developmental disorder in Down’s syndrome. J Autism Dev Disord. 2007;37:1394–5.
de la Chapelle A, Herva R, Koivisto M, Aula P. A deletion in chromosome 22 can cause DiGeorge syndrome. Hum Genet. 1981;57:253–6.
Kelley RI, Zackai EH, Emanuel BS, Kistenmacher M, Greenberg F, Punnett HH. The association of the DiGeorge anomalad with partial monosomy of chromosome 22. J Pediatr. 1982;101:197–200.
Bassett AS, Chow EW, Husted J, Weksberg R, Caluseriu O, Webb GD, et al. Clinical features of 78 adults with 22q11 deletion syndrome. Am J Med Genet A. 2005;138:307–13.
Carey AH, Kelly D, Halford S, Wadey R, Wilson D, Goodship J, et al. Molecular genetic study of the frequency of monosomy 22q11 in DiGeorge syndrome. Am J Hum Genet. 1992;51:964–70.
• Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, et al. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43:838–46. Evaluates the pathogenicity of several CNVs by comparing their frequency in cases referred for clinical genetic testing and control individuals.
Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–9.
•• Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–85. Assessed the role of CNVs in a large cohort of people with autism and identified several high risk de novo CNVs, including a duplication of 7q11.23.
Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet. 2014;94:677–94.
Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM, et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–43.
Levinson DF, Duan J, Oh S, Wang K, Sanders AR, Shi J, et al. Copy number variants in schizophrenia: confirmation of five previous findings and new evidence for 3q29 microdeletions and VIPR2 duplications. Am J Psychiatry. 2011;168:302–16.
Mefford HC, Sharp AJ, Baker C, Itsara A, Jiang Z, Buysse K, et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N Engl J Med. 2008;359:1685–1699.
Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358:667–75.
Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, Badner JA, et al. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet. 2008;17:628–38.
Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–88.
McCarthy SE, Makarov V, Kirov G, Addington AM, McClellan J, Yoon S, et al. Microduplications of 16p11.2 are associated with schizophrenia. Nat Genet. 2009;41:1223–7.
Moreno-De-Luca D, Mulle JG, Kaminsky EB, Sanders SJ, Myers SM, Adam MP, et al. Deletion 17q12 is a recurrent copy number variant that confers high risk of autism and schizophrenia. Am J Hum Genet. 2010;87:618–30.
Liu P, Carvalho CM, Hastings PJ, Lupski JR. Mechanisms for recurrent and complex human genomic rearrangements. Curr Opin Genet Dev. 2012;22:211–20.
•• Stefansson H, Meyer-Lindenberg A, Steinberg S, Magnusdottir B, Morgen K, Arnarsdottir S, et al. CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature. 2014;505:361–6. Demonstrates that control subjects who have CNVs previously associated with neuropsychiatric disorders have a cognitive functioning that falls between affected individuals and population controls.
Klei L, Sanders SJ, Murtha MT, Hus V, Lowe JK, Willsey AJ, et al. Common genetic variants, acting additively, are a major source of risk for autism. Mol Autism. 2012;3:9.
• Szatkiewicz JP, O’Dushlaine C, Chen G, Chambert K, Moran JL, Neale BM, et al. Copy number variation in schizophrenia in Sweden. Mol Psychiatry. 2014. Confirms the association of several recurrent CNVs with schizophrenia and identifies additional CNVs conferring risk for this disorder in a large sample of Swedish individuals.
Botto LD, May K, Fernhoff PM, Correa A, Coleman K, Rasmussen SA, et al. A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics. 2003;112:101–7.
DiGeorge AM. Congenital absence of the thymus and its immunologic consequences: concurrence with congenital hypoparathyroidism. Birth Defects. 1968;4:116–21.
Kinouchi A, Mori K, Ando M, Takao A. Facial appearance of patients with conotruncal anomalies. Pediatr Jpn. 1976;17:84.
Shprintzen RJ, Goldberg RB, Lewin ML, Sidoti EJ, Berkman MD, Argamaso RV, et al. A new syndrome involving cleft palate, cardiac anomalies, typical facies, and learning disabilities: velo-cardio-facial syndrome. Cleft Palate J. 1978;15:56–62.
Scambler PJ, Carey AH, Wyse RK, Roach S, Dumanski JP, Nordenskjold M, et al. Microdeletions within 22q11 associated with sporadic and familial DiGeorge syndrome. Genomics. 1991;10:201–6.
Driscoll DA, Budarf ML, Emanuel BS. A genetic etiology for DiGeorge syndrome: consistent deletions and microdeletions of 22q11. Am J Hum Genet. 1992;50:924–33.
Burn J, Takao A, Wilson D, Cross I, Momma K, Wadey R, et al. Conotruncal anomaly face syndrome is associated with a deletion within chromosome 22q11. J Med Genet. 1993;30:822–4.
Scambler PJ, Kelly D, Lindsay E, Williamson R, Goldberg R, Shprintzen R, et al. Velo-cardio-facial syndrome associated with chromosome 22 deletions encompassing the DiGeorge locus. Lancet. 1992;339:1138–9.
Schneider M, Debbané M, Bassett AS, Chow EW, Fung WL, van den Bree MB, et al. Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: results from the International Consortium on brain and behavior in 22q11.2 deletion syndrome. Am J Psychiatry. 2014;171:627–39.
Wentzel C, Fernström M, Ohrner Y, Annerén G, Thuresson AC. Clinical variability of the 22q11.2 duplication syndrome. Eur J Med Genet. 2008;51:501–10.
Steffenburg S, Gillberg CL, Steffenburg U, Kyllerman M. Autism in Angelman syndrome: a population-based study. Pediatr Neurol. 1996;14:131–6.
Petersen MB, Brondum-Nielsen K, Hansen LK, Wulff K. Clinical, cytogenetic, and molecular diagnosis of Angelman syndrome: estimated prevalence rate in a Danish county. Am J Med Genet. 1995;60:261–2.
Angelman H. ‘Puppet’ children a report on three cases. Dev Med Child Neurol. 1965;7:681–8.
Trillingsgaard A, Ostergaard JR. Autism in Angelman syndrome: an exploration of comorbidity. Autism. 2004;8:163–74.
Kaplan LC, Wharton R, Elias E, Mandell F, Donlon T, Latt SA. Clinical heterogeneity associated with deletions in the long arm of chromosome 15: report of 3 new cases and their possible genetic significance. Am J Med Genet. 1987;28:45–53.
Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet. 1997;15:70–3.
Matsuura T, Sutcliffe JS, Fang P, Galjaard RJ, Jiang YH, Benton CS, et al. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat Genet. 1997;15:74–7.
Beaudet AL. Angelman syndrome: drugs to awaken a paternal gene. Nature. 2012;481:150–2.
Whittington JE, Holland AJ, Webb T, Butler J, Clarke D, Boer H. Population prevalence and estimated birth incidence and mortality rate for people with Prader–Willi syndrome in one UK Health Region. J Med Genet. 2001;38:792–8.
Prader A, Labhart A, Willi H. Ein Syndrom von Adipositas, Kleinwuchs, Kryptorchismus und Oligophrenie nach Myatonieartigem Zustand im Neugeborenenalter. Schweiz Med Wschr. 1965;86:1260–1.
Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader–Willi syndrome. Genet Med. 2012;14:10–26.
Dykens EM, Lee E, Roof E. Prader–Willi syndrome and autism spectrum disorders: an evolving story. J Neurodev Disord. 2011;3:225–37.
Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawford JD. Deletions of chromosome 15 as a cause of the Prader–Willi syndrome. N Engl J Med. 1981;304:325–9.
Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, et al. Prader–Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet. 2008;40:719–21.
Depienne C, Moreno-De-Luca D, Heron D, Bouteiller D, Gennetier A, Delorme R, et al. Screening for genomic rearrangements and methylation abnormalities of the 15q11-q13 region in autism spectrum disorders. Biol Psychiatry. 2009;66:349–59.
Buiting K. Prader–Willi syndrome and Angelman syndrome. Am J Med Genet C Semin Med Genet. 2010;154C:365–76.
Gillberg C, Steffenburg S, Wahlström J, Gillberg IC, Sjöstedt A, Martinsson T, et al. Autism associated with marker chromosome. J Am Acad Child Adolesc Psychiatry. 1991;30:489–94.
Callen DF, Eyre H, Yip MY, Freemantle J, Haan EA. Molecular cytogenetic and clinical studies of 42 patients with marker chromosomes. Am J Med Genet. 1992;43:709–15.
Cook EH, Lindgren V, Leventhal BL, Courchesne R, Lincoln A, Shulman C, et al. Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am J Hum Genet. 1997;60:928–34.
Battaglia A. The inv dup (15) or idic (15) syndrome (Tetrasomy 15q). Orphanet J Rare Dis. 2008;3:30.
Christian SL, Brune CW, Sudi J, Kumar RA, Liu S, Karamohamed S, et al. Novel submicroscopic chromosomal abnormalities detected in autism spectrum disorder. Biol Psychiatry. 2008;63:1111–7.
Tucker T, Giroux S, Clément V, Langlois S, Friedman JM, Rousseau F. Prevalence of selected genomic deletions and duplications in a French–Canadian population-based sample of newborns. Mol Genet Genomic Med. 2013;1:87–97.
Shinawi M, Liu P, Kang SH, Shen J, Belmont JW, Scott DA, et al. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J Med Genet. 2010;47:332–41.
• Zufferey F, Sherr EH, Beckmann ND, Hanson E, Maillard AM, Hippolyte L, et al. A 600 kb deletion syndrome at 16p11.2 leads to energy imbalance and neuropsychiatric disorders. J Med Genet. 2012;49:660–8. Shows that full scale intelligence quotient of people with 16p11.2 deletions is two standard deviations lower than control individuals from their families, and that obesity is present in 50 s% of people with this CNV but it is not correlated with IQ or any behavioral trait.
Fernandez BA, Roberts W, Chung B, Weksberg R, Meyn S, Szatmari P, et al. Phenotypic spectrum associated with de novo and inherited deletions and duplications at 16p11.2 in individuals ascertained for diagnosis of autism spectrum disorder. J Med Genet. 2010;47:195–203.
Jacquemont S, Reymond A, Zufferey F, Harewood L, Walters RG, Kutalik Z, et al. Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature. 2011;478:97–102.
Williams JC, Barratt-Boyes BG, Lowe JB. Supravalvular aortic stenosis. Circulation. 1961;24:1311–8.
Beuren AJ, Apitz J, Harmjanz D. Supravalvular aortic stenosis in association with mental retardation and a certain facial appearance. Circulation. 1962;26:1235–40.
Pober BR. Williams–Beuren syndrome. N Engl J Med. 2010;362:239–52.
Ewart AK, Morris CA, Atkinson D, Jin W, Sternes K, Spallone P, et al. Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat Genet. 1993;5:11–6.
Tordjman S, Anderson GM, Cohen D, Kermarrec S, Carlier M, Touitou Y, et al. Presence of autism, hyperserotonemia, and severe expressive language impairment in Williams–Beuren syndrome. Mol Autism. 2013;4:29.
Somerville MJ, Mervis CB, Young EJ, Seo EJ, del Campo M, Bamforth S, et al. Severe expressive-language delay related to duplication of the Williams–Beuren locus. N Engl J Med. 2005;353:1694–701.
Berg JS, Brunetti-Pierri N, Peters SU, Kang S-HL, Fong C-T, Salamone J, et al. Speech delay and autism spectrum behaviors are frequently associated with duplication of the 7q11.23 Williams–Beuren syndrome region. Genet Med. 2007;9:427–41.
Van der Aa N, Rooms L, Vandeweyer G, van den Ende J, Reyniers E, Fichera M, et al. Fourteen new cases contribute to the characterization of the 7q11.23 microduplication syndrome. Eur J Med Genet. 2009;52:94–100.
Mulle JG, Pulver AE, McGrath JA, Wolyniec PS, Dodd AF, Cutler DJ, et al. Reciprocal duplication of the Williams–Beuren syndrome deletion on chromosome 7q11.23 is associated with schizophrenia. Biol Psychiatry. 2014;75:371–7.
Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–41.
He X, Sanders SJ, Liu L, De Rubeis S, Lim ET, Sutcliffe JS, et al. Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet. 2013;9:e1003671.
Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol. 2007;17:103–11.
Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell. 2013;155:997–1007.
Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013;155:1008–21.
Uddin M, Tammimies K, Pellecchia G, Alipanahi B, Hu P, Wang Z, et al. Brain-expressed exons under purifying selection are enriched for de novo mutations in autism spectrum disorder. Nat Genet. 2014;46:742–7.
Gulsuner S, Walsh T, Watts AC, Lee MK, Thornton AM, Casadei S, et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell. 2013;154:518–29.
•• Moreno-De-Luca A, Myers SM, Challman TD, Moreno-De-Luca D, Evans DW, Ledbetter DH. Developmental brain dysfunction: revival and expansion of old concepts based on new genetic evidence. Lancet Neurol. 2013;12:406–14. Highlights the variability of the clinical features observed in people with the same genetic variants, spanning across different diagnostic boundaries, and proposes the concept of developmental brain dysfunction to capture this phenotypic diversity.
Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, et al. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43(9):838–46.
•• Moreno-De-Luca D, Sanders S, Willsey A, Mulle J, Lowe J, Geschwind D, et al. Using large clinical datasets to infer pathogenicity for rare copy number variants in autism cohorts. Mol Psychiatry. 2012. Uses a tiered approach to identify pathogenic CNVs, initially deemed to have deleterious effects in large clinical cohorts, in autism cohorts that would have otherwise been to small to establish those CNVs as conferring high risk for autism.
Stahlberg O, Soderstrom H, Rastam M, Gillberg C. Bipolar disorder, schizophrenia, and other psychotic disorders in adults with childhood onset AD/HD and/or autism spectrum disorders. J Neural Transm. 2004;111:891–902.
Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–84.
Skuse DH. Rethinking the nature of genetic vulnerability to autistic spectrum disorders. Trends Genet. 2007;23:387–95.
Rees E, Kirov G, Sanders A, Walters JT, Chambert KD, Shi J, et al. Evidence that duplications of 22q11.2 protect against schizophrenia. Mol Psychiatry. 2014;19:37–40.
de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367:1921–9.
Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–82.
Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485(7397):242–5.
Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, et al. De Novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–99.
O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485(7397):246–50.
Arvio M, Sillanpää M. Prevalence, aetiology and comorbidity of severe and profound intellectual disability in Finland. J Intellect Disabil Res. 2003;47:108–12.
Reddy KS. Cytogenetic abnormalities and fragile-X syndrome in autism spectrum disorder. BMC Med Genet. 2005;6:3.
Shen Y, Dies KA, Holm IA, Bridgemohan C, Sobeih MM, Caronna EB, et al. Clinical genetic testing for patients with autism spectrum disorders. Pediatrics. 2010;125:e727–35.
Acknowledgments
Our sincere thanks to C. L. Martin and M. W. State for their insightful comments during the preparation of this manuscript. In writing this review we were struck by how many of researchers that laid the bedrock of the current understanding of the genetics of neuropsychiatric disorders continue to dedicate themselves to this task today. We value their support and mentorship all the more for better understanding their contribution. This work was supported by the Howard Hughes Medical Institute (International Student Research Fellowship to S.J.S.), the National Institute of Mental Health (5R25MH071584-07 and 5T32MH19961-14 to D. M., R01-MH101720, R01-MH071537 and P50-MH100023 to J.F.C.), the Georgia Department of Behavioral Health and Developmental Disabilities (J.F.C.), and the Simons Foundation (J.F.C. and S.J.S.).
Disclosure
D. Moreno-De-Luca, A. Moreno-De-Luca, J. F. Cubells, and S. J. Sanders declare no conflicts of interest.
Human and Animal Rights and Informed Consent
All studies by D Moreno-De-Luca, A Moreno-De-Luca, JF Cubells, and SJ Sanders involving animal and/or human subjects were performed after approval by the appropriate institutional review boards. When required, written informed consent was obtained from all participants.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Moreno-De-Luca, D., Moreno-De-Luca, A., Cubells, J.F. et al. Cross-Disorder Comparison of Four Neuropsychiatric CNV Loci. Curr Genet Med Rep 2, 151–161 (2014). https://doi.org/10.1007/s40142-014-0045-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40142-014-0045-7