
Abstract
The GATA2 gene codes for a master hematopoietic transcription factor that is essential for the proliferation and maintenance of hematopoietic stem and progenitor cells. Heterozygous germline mutations in GATA2 have been initially associated with several clinical entities that are now collectively defined as GATA2 deficiency. Despite pleiotropic clinical manifestations, the high propensity for the development of myelodysplastic syndromes (MDS) constitutes the most common clinical denominator of this major MDS predisposition syndrome. The immunological phenotypes can be variable and mostly include deficiency of monocytes and/or B cells. Thus far, nearly 380 GATA2-deficient patients had been reported, with a roughly estimated prevalence of myeloid neoplasia of at least 75%. The most common abnormal karyotypes associated with GATA2-related MDS are monosomy 7, der(1;7) and trisomy 8. The overall clinical penetrance seems to be nearly complete for this transcriptopathy disorder. The high-risk MDS subtypes and karyotypes, and the underlying immunodeficiency guide decision-making toward timely stem cell transplantation.
Similar content being viewed by others
Introduction
Historically, familial myelodysplastic syndromes (MDS)/acute myeloid leukemia (AML) with nonsyndromic manifestation have been occasionally described in association with monosomy 7 karyotypes [1]. Germline mutations in genes coding for transcription factors CEBPA and RUNX1 were discovered as the cause of autosomal dominant familial MDS/AML syndromes, but the genetic cause remained obscure in many reported pedigrees. In 2011, loss-of-function (LOF) mutations or deletions in the GATA2 gene were identified as the third major MDS/AML predisposition syndrome [2]. Strikingly, at almost same time, germline GATA2 mutations were brought in association with other clinical entities, namely the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome [3], dendritic cell, monocyte, B and NK lymphoid (DCML) deficiency [4] and primary lymphedema associated with predisposition to acute myeloid leukemia (Emberger syndrome) [5]. Finally, GATA2 deficiency had been identified as the most common known genetic cause of primary childhood MDS, where the majority of affected cases had negative family history [6]. Today, it is well accepted that all these clinical manifestations belong to the broad spectrum of a single genetic disease.
Patients with inherited bone marrow failure disorders (IBMFS) such as Fanconi anemia and severe congenital neutropenia have an increased risk of MDS/AML, and a differential diagnosis of IBMFS is important for treatment stratification [7]. From this point of view, the exclusion of germline GATA2 mutations and other familial MDS/AML predisposition syndromes clearly also has an impact on therapy recommendations and family counseling. These aspects are now addressed in a new chapter on myeloid neoplasms with germ line predisposition, included in the 2016 revision of the WHO classification of hematological malignancies [8].
In this review, we summarize key information on myeloid neoplasms originating from germline GATA2 mutation.
Role of GATA2 in normal and malignant hematopoiesis
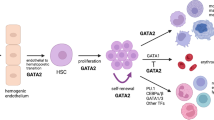
The GATA2 gene encodes a chief hematopoietic transcription factor that through its 2 zinc finger domains (ZF) can occupy GATA DNA motifs in several thousand genes [9]. While ZF1 is thought to be involved in protein–protein interaction, ZF2 can control transcription through protein-DNA binding. GATA2 plays a critical role in hematopoietic development as it controls the transition from hemogenic endothelium to hematopoietic stem cells (HCS) and is required for HSC survival and self-renewal, through cooperative processes involving other transcription factors (Fig. 1) [9]. GATA2 itself cooperates in a complex network of transcription factors (TF), and depending on the stage of hematopoiesis it can be activated or repressed by other TF, as well as control the expression of other TF crucial for lineage development. Homozygous Gata2 knock-out mice exhibit embryonic lethality due to the failure of definitive hematopoiesis [10]. Heterozygous knock-out Gata2 mice demonstrate compromised HSC longevity, leading to reduced numbers of progenitor cells, with no occurrence of MDS or leukemia [11].

GATA2 and related transcription factors in hematopoietic development. GATA2 plays a pivotal role in emergence of hematopoietic stem cells from hemogenic endothelium in the process called endothelial to hematopoietic transition (ETH). In this process GATA2 expression is regulated by NOTCH1 and BMP4, interacting with other hematopoietic players, involving FLI1, SCL/TAL1 and RUNX1. Additionally, the HSC proliferation is controlled by EVI1, which binds to the GATA2 promoter as an enhancer. While in monocytic and erythroid development, GATA2 expression is switched off or displaced by other transcription factors, GATA2 is involved in the differentiation of mast cells and megakaryocytes
Genetic causes of GATA2 deficiency
The GATA2 gene is located the long arm of chromosome 3 at position 21.3. Retrospectively, the first report likely depicting GATA2 deficiency in context of a large interstitial microdeletion (3q21.1-q21.3) was reported in 2008 [12]. Three major types of monoallelic mutations are known to cause GATA2 deficiency: truncating or splice site mutations leading to premature translation termination prior or within ZF2, missense mutations within ZF2, and noncoding variants in the +9.5 kb enhancer region of intron 4. In addition, small in frame or whole gene deletions are encountered on single occasions. The typical mutational landscape in an exemplary cohort of patients with GATA2-related pediatric MDS [8] is depicted in Fig. 2. Overall, the mutations seem to result in haploinsufficiency. LOF-effect was demonstrated for the ZF2 mutations Thr354Met and Thr355del found in familial MDS/AML [2]; Arg361Leu and Cys373Arg in Emberger syndrome [5]. Noncoding GATA2 variants in intronic regulatory region leading to haploinsufficiency were initially reported by Holland's group [13, 14] in the +9.5 intronic region that acts as GATA2 transcriptional enhancer [15]. Notably, 10.5% of patients with pediatric MDS and GATA2 mutation carried these noncoding variants (Fig. 2). The term GATA2 deficiency or haploinsufficiency has been widely accepted to describe GATA2-related disorders. On the other hand, somatic GATA2 mutations in adult AML can occur in both ZF regions with preference for ZF1 [16, 17] and can exhibit both loss- or gain-of-function effects [16, 18, 19]. Generally, only one GATA2 allele is affected in carriers and (unlike for DDX41 or RUNX1) secondary somatic GATA2 mutations are not detected. However, in one study, two heterozygous mutations Thr358Asn and Leu359Val were identified in-cis in an individual from a family presenting MDS/AML [20]. Interestingly, Leu359Val mutation displayed significant gain of function [21]. The current diagnostic workup for suspected GATA2 deficiency should include Sanger- or NGS-based analysis of the coding sequence, intron 4 enhancer, and copy number analysis to rule out GATA2 gene deletion. It is important to note that for the confirmation of germline status, skin fibroblasts or hair follicles offer the optimal source for germline DNA, while the use of buccal swabs might not be valid due to contamination with blood leukocytes (and in case of a positive result does not prove if the mutation is truly germline).

GATA2 germline mutations in children and adolescents with MDS. Structure of GATA2 protein with two functionally important zinc finger (ZF) domains marked in green (ZF1) and red (ZF2). 42 germline GATA2 mutations are depicted, as previously reported by Wlodarski et al. [6]. Red-color circles represent affected amino acids. Numbers in brackets indicate the numbers of cases with a particular variant. Bold font denotes nonsense mutations, whereas bold italic font demonstrate splice site mutations
Clinical phenotype of GATA2 deficiency
Long-term observation of patients and family carriers in several patient cohorts allows to construct a provisional model of GATA2 disease evolution (Fig. 3). After uneventful pregnancy, birth and preschool years, GATA2-mutation carriers can develop progressive cellular deficiency (e.g., of monocytes, B-, NK-cells) and cytopenias (Fig. 3; Table 1). This can continue for a few to tens of years and eventually, in the adulthood culminate in leukemic progress with the development of AML or other type of myeloid neoplasia. Notably, in children, the disease can take an "alternative" course where MDS can develop all of a sudden after unremarkable clinical history. However, preexisting immunodeficiency was shown to be retrospectively present in at least 50% of GATA2-carriers who developed MDS, pointing to the possibility of preexisting immuno-cytopenias might be present in more patients [6]. Depending on the studied patient cohort, the median age at diagnosis for roughly 380 cases in the literature was shown to be in range from 12 to 35 years (average 19.7 years), with 75% of carriers developing myeloid neoplasia [22]. The most common karyotypes were monosomy 7 or der(1;7) found in 41% of all carriers (with predilection for pediatric cohorts where 75–80% of cases acquire this karyotype), and trisomy 8 detected on average in 15% of reported cases [22]. It is worth to remark that del5q and complex karyotypes are generally not encountered in GATA2 deficiency.

Evolution of MDS in GATA2-deficient background. As a result of GATA2 haploinsufficiency, stem cell exhaustion can develop over the course of decades and result in cellular deficiencies defining the immunodeficiency phenotype. Additionally, either as an independent clinical phenotype, or during later disease stages following hypocellular stage, myeloid neoplasia can develop and be associated with accumulation of specific cytogenetic abnormalities or somatic mutations. Bone marrow cellularity in GATA2-deficient bone marrow likely changes according to disease stage
Dysmorphic features can be present in some but not all affected patients, and include congenital deafness, facial anomalies, urogenital malformations; behavioral problems such as autism spectrum disorders can be also present. Other non-hematologic disease manifestation is variable: some patients may develop pulmonary problems, autoimmune symptoms (e.g., lupus-like disease, immune cytopenias), thrombosis, and generalized warts and HPV-related cancers (Table 1). In summary, hematopoietic, immune, lymphatic, vascular, urogenital, and neurological systems can be affected by GATA2 deficiency. The overall penetrance for hematologic malignancy is very high, and for the occurrence of any of the specific disease symptoms is nearly complete.
MonoMAC syndrome/DCML deficiency
Immunodeficiency was the leading medical problem in the initial cohorts described with GATA2 mutations: autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome [3], dendritic cell, monocyte, B and NK lymphoid (DCML) deficiency [4] with predisposition to MDS/AML. Generalized warts related to HPV infection were common viral complication, in addition to severe HSV, VZV, EBV and CMV infections. Disseminated non-tuberculous mycobacteria (NTM) infections were observed in about half of cases with MonoMAC syndrome [23]. Severe bacterial or fungal infections, and pulmonary alveolar proteinosis (PAP) were also prevalent in MonoMAC syndrome. Notably, after HSCT, the rates of infections and PAP can significantly decrease [24].
Emberger syndrome
The association of primary lymphedema and predisposition to MDS/AML with or without congenital deafness by autosomal dominant inheritance was reported by Emberger et al. [25]. Germline GATA2 mutations were identified as a single common denominator by whole exome sequencing [5]. GATA2 protein is expressed at high levels in endothelial cells and lymphatic vessel valves [26, 27] and controls the expression of PROX1 and FOXC2 genes important for programming lymphatic valve development [28]. In one study, it has been suggested that N-terminal frameshift mutations or larger deletions of GATA2 are more likely to cause lymphedema [23]; however, this association could not be confirmed in other patient cohorts [6]. Congenital deafness is presumed to result from failure of generation of the perilymphatic space surrounding the semicircular ducts in inner ear [29].
Familial MDS/AML
Germline GATA2 mutations were detected in four pedigrees with an autosomal dominant inheritance pattern of MDS/AML in 2011 [2]. Variable clinical manifestations were shown in large pedigrees [30]. It is not understood, however, why some patients develop MDS while carriers of identical mutations in the same family do not develop relevant hematologic symptoms.
Pediatric MDS
Our group has screened more than 600 cases of primary or secondary MDS in children and adolescents who were enrolled in the European Working Group of MDS in childhood. The overall frequency of germline GATA2 mutations was 15% for advanced and 7% for all primary MDS cases. Surprisingly, 72% of adolescents diagnosed with MDS and monosomy 7 harbor germline mutations in GATA2. Conversely, mutations were absent in the group with secondary MDS that was therapy related or occurred after aplastic anemia. We propose that GATA2 screening should be included in the workup of all children and young adults with monosomy 7, trisomy 8, or independent of karyotype if presenting with features suspicious for GATA2 deficiency [6, 31]. Interestingly, in pediatric MDS cohorts, not monocytopenia but B-cell lymphopenia (including progenitors in bone marrow) was identified as the most consistent immunological feature [32, 33]. This might be due to the fact that true functional monocytopenia might be masked by the expansion of malignant myelo-monocytic lineage in the setting of MDS with monosomy 7.
CMML/JMML
Monocytopenia has been previously proposed as a diagnostic feature of GATA2 deficiency. However, some patients with GATA2-related MDS can present with monocytosis rather than monocytopenia [6, 34]. Somatic ASXL1 mutations are associated with the presence of monosomy 7, BM hypercellularity and CMML [35]. Furthermore, several (rare) cases of adult CMML disease were reported in germline GATA2 mutation carriers. GATA2 deficiency seems not to play a role in the pathogenesis of JMML [36].
Aplastic anemia
GATA2 deficiency-associated bone marrow disorder can present with features overlapping with aplastic anemia. Distinguishing GATA2 patients from aplastic anemia is critical for selecting appropriate therapy. Four out of 32 patients with suspected aplastic anemia who had features suspicious for GATA2 mutations were identified by DNA sequencing [37].
Chronic neutropenia
The analysis of patients enrolled in the French Severe Chronic Neutropenia Registry identified 7 pedigrees with germline GATA2 mutations who presented with mild chronic neutropenia associated with immunodeficiency and subsequent MDS evolution [38].
Acquired genetic abnormalities in carriers of germline GATA2 mutations
The mechanism of malignant clonal evolution in GATA2-deficient patients is not understood. First, recurrent karyotype abnormalities involving chromosomes 7 and 8 point to their mechanistic relevance in context of underlying GATA2 deficiency. However, these cytogenetic aberrations are not specific to GATA2 deficiency, as they also can arise in MDS originating from hereditary predisposition syndromes [39]. Second, recurrent loss-of-function ASXL1 mutations have been described in patients with GATA2 deficiency. However, the presence of ASXL1 mutations seems to be determined by the underlying karyotype, namely monosomy 7 [40]. This most frequent karyotype in pediatric is associated with ASXL1 and SETBP1 mutations, independently of germline GATA2 mutational status [34, 41]. Similarly to ASXL1, the presence of somatic SETBP1 mutations in GATA2-related MDS seems to be associated with monosomy 7 [42]. From functional perspective, the association of SETBP1-ASXL1 mutations was postulated as a cooperative mechanism advancing leukemic transformation [43]. Finally, recurrent mutations in STAG2 gene were identified in several cases with GATA2 deficiency; however, the functional significance is not known [41, 44, 45].
Treatment of myeloid neoplasms with germline GATA2 mutations
Because the phenotypic heterogeneity is not only evident between different non-related carriers of the same mutation, but also within a single family, it is difficult giving advice on the individual outcomes and recommending tailored treatment strategies. Nevertheless, early diagnosis of GATA2 deficiency can help to avoid unnecessary or toxic therapies, for example, prolonged immunosuppression or AML-type chemotherapy given for advanced MDS. Overall, non-curative therapies should be limited and because of the high risk for evolution of advanced MDS with unfavorable karyotypes, timely HSCT should be suggested as a curative approach. Overall survival (OS) of GATA2-mutated patients transplanted for immunodeficiency was shown to be 54% at 4 years after transplantation in a NIH-based study [23]. In pediatric GATA2 cohorts, 5-year OS was 66% in patients transplanted for MDS with monosomy 7. Notably, OS and outcome of HSCT were not influenced by GATA2 mutational status [6]. Our findings indicate that GATA2 deficiency itself does not increase transplant-related mortality in affected children (most of the patients received myeloablative regimen due to monosomy 7). Ideally, HSCT should be performed before the development of MDS with excess of blasts, cytogenetic clone, and oncogenic somatic mutations. These factors support the need for a close monitoring of GATA2 mutation carriers for the occurrence of any of these events.
Concluding remarks
GATA2 deficiency belongs to the disease group of transcriptopathies predisposing to myeloid neoplasia. The heterogenous clinical manifestation, ranging from immunodeficiency, vascular phenotypes, deafness, to sporadic myeloid neoplasia illustrates the pleiotropic function of this master transcription factor. However, many important questions remain unanswered at present. For example, it is not known what drives the development of delayed onset myeloid neoplasia in GATA2-haploinsufficient background. Among many hypotheses, one could speculate that either a chronic pathogen challenge might be toxic to the BM and result in leukemic transformation, or GATA2 deficiency itself results in dose perturbation of other transcription factors such as RUNX1 or PU1 which themselves can act as oncogenes. Another question is what determines the variable clinical expressivity where in one family several affected carriers display varying phenotypes and develop MDS at different age. Thus far, there is no evidence of revertant somatic mosaicism (such as encountered in Fanconi anemia) in GATA2 deficiency; however, other mechanisms that control the rate of allelic expression might be the cause, e.g., epigenetic modulation. Several of these questions should be answered in prospective international studies.
References
Owen C, Barnett M, Fitzgibbon J. Familial myelodysplasia and acute myeloid leukaemia—a review. Br J Haematol. 2008;140:123–32.
Hahn CN, Chong CE, Carmichael CL, Wilkins EJ, Brautigan PJ, Li XC, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet. 2011;43:1012–7.
Hsu AP, Sampaio EP, Khan J, Calvo KR, Lemieux JE, Patel SY, et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood. 2011;118:2653–5.
Dickinson RE, Griffin H, Bigley V, Reynard LN, Hussain R, Haniffa M, et al. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood. 2011;118:2656–8.
Ostergaard P, Simpson MA, Connell FC, Steward CG, Brice G, Woollard WJ, et al. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nat Genet. 2011;43:929–31.
Wlodarski MW, Hirabayashi S, Pastor V, Stary J, Hasle H, Masetti R, et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood. 2016;127:1387–97.
Hasle H, Niemeyer CM. Advances in the prognostication and management of advanced MDS in children. Br J Haematol. 2011;154:185–95.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405.
Dore LC, Chlon TM, Brown CD, White KP, Crispino JD. Chromatin occupancy analysis reveals genome-wide GATA factor switching during hematopoiesis. Blood. 2012;119:3724–33.
Tsai FY, Keller G, Kuo FC, Weiss M, Chen J, Rosenblatt M, et al. An early haematopoietic defect in mice lacking the transcription factor GATA-2. Nature. 1994;371:221–6.
Rodrigues NP, Janzen V, Forkert R, Dombkowski DM, Boyd AS, Orkin SH, et al. Haploinsufficiency of GATA-2 perturbs adult hematopoietic stem-cell homeostasis. Blood. 2005;106:477–84.
Callier P, Faivre L, Marle N, Thauvin-Robinet C, Guy J, Mosca AL, et al. Detection of an interstitial 3q21.1-q21.3 deletion in a child with multiple congenital abnormalities, mental retardation, pancytopenia, and myelodysplasia. Am J Med Genet A. 2009;149A:1323–6.
Hsu AP, Johnson KD, Falcone EL, Sanalkumar R, Sanchez L, Hickstein DD, et al. GATA2 haploinsufficiency caused by mutations in a conserved intronic element leads to MonoMAC syndrome. Blood. 2013;121(3830–7):S1–7.
Johnson KD, Hsu AP, Ryu MJ, Wang J, Gao X, Boyer ME, et al. Cis-element mutated in GATA2-dependent immunodeficiency governs hematopoiesis and vascular integrity. J Clin Investig. 2012;122:3692–704.
Wozniak RJ, Boyer ME, Grass JA, Lee Y, Bresnick EH. Context-dependent GATA factor function: combinatorial requirements for transcriptional control in hematopoietic and endothelial cells. J Biol Chem. 2007;282:14665–74.
Zhang SJ, Ma LY, Huang QH, Li G, Gu BW, Gao XD, et al. Gain-of-function mutation of GATA-2 in acute myeloid transformation of chronic myeloid leukemia. Proc Natl Acad Sci USA. 2008;105:2076–81.
Hou HA, Lin YC, Kuo YY, Chou WC, Lin CC, Liu CY, et al. GATA2 mutations in patients with acute myeloid leukemia-paired samples analyses show that the mutation is unstable during disease evolution. Ann Hematol. 2015;94:211–21.
Celton M, Forest A, Gosse G, Lemieux S, Hebert J, Sauvageau G, et al. Epigenetic regulation of GATA2 and its impact on normal karyotype acute myeloid leukemia. Leukemia. 2014;28:1617–26.
Greif PA, Dufour A, Konstandin NP, Ksienzyk B, Zellmeier E, Tizazu B, et al. GATA2 zinc finger 1 mutations associated with biallelic CEBPA mutations define a unique genetic entity of acute myeloid leukemia. Blood. 2012;120:395–403.
Gao J, Gentzler RD, Timms AE, Horwitz MS, Frankfurt O, Altman JK, et al. Heritable GATA2 mutations associated with familial AML-MDS: a case report and review of literature. J Hematol Oncol. 2014;7:36.
Hahn CN, Brautigan PJ, Chong CE, Janssan A, Venugopal P, Lee Y, et al. Characterisation of a compound in-cis GATA2 germline mutation in a pedigree presenting with myelodysplastic syndrome/acute myeloid leukemia with concurrent thrombocytopenia. Leukemia. 2015;29:1795–7.
Wlodarski M, Collin M, Horwitz MS. GATA2 deficiency and related myeloid neoplasms. Semin Hematol. 2017;54(2):81–86.
Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, Calvo KR, et al. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood. 2014;123:809–21.
Cuellar-Rodriguez J, Gea-Banacloche J, Freeman AF, Hsu AP, Zerbe CS, Calvo KR, et al. Successful allogeneic hematopoietic stem cell transplantation for GATA2 deficiency. Blood. 2011;118:3715–20.
Emberger JM, Navarro M, Dejean M, Izarn P. Deaf-mutism, lymphedema of the lower limbs and hematological abnormalities (acute leukemia, cytopenia) with autosomal dominant transmission. J Genet Hum. 1979;27:237–45.
Kazenwadel J, Secker GA, Liu YJ, Rosenfeld JA, Wildin RS, Cuellar-Rodriguez J, et al. Loss-of-function germline GATA2 mutations in patients with MDS/AML or MonoMAC syndrome and primary lymphedema reveal a key role for GATA2 in the lymphatic vasculature. Blood. 2012;119:1283–91.
Lim KC, Hosoya T, Brandt W, Ku CJ, Hosoya-Ohmura S, Camper SA, et al. Conditional Gata2 inactivation results in HSC loss and lymphatic mispatterning. J Clin Investig. 2012;122:3705–17.
Kazenwadel J, Betterman KL, Chong CE, Stokes PH, Lee YK, Secker GA, et al. GATA2 is required for lymphatic vessel valve development and maintenance. J Clin Investig. 2015;125:2979–94.
Haugas M, Lillevali K, Hakanen J, Salminen M. Gata2 is required for the development of inner ear semicircular ducts and the surrounding perilymphatic space. Dev Dyn. 2010;239:2452–69.
Mutsaers PG, van de Loosdrecht AA, Tawana K, Bodor C, Fitzgibbon J, Menko FH. Highly variable clinical manifestations in a large family with a novel GATA2 mutation. Leukemia. 2013;27:2247–8.
Hirabayashi S, Strahm B, Urbaniak S, Karow A, Cseh A, van den Heuvel-Eibrink M, et al. Unexpected high frequency of GATA2 mutations in children with non-familial MDS and monosomy 7. Blood. 2012;120(21):1699 (abstract).
Novakova M, Janda A, Wlodarski MW, Sukova M, Campr V, Zaliova MK, et al. Defect in B cell production driven by GATA2 mutation results in their absolute reduction and mature phenotype in pediatric patients. Blood. 2014;124(21):2746 (abstract).
Novakova M, Zaliova M, Sukova M, Wlodarski M, Janda A, Fronkova E, et al. Loss of B cells and their precursors is the most constant feature of GATA-2 deficiency in childhood myelodysplastic syndrome. Haematologica. 2016;101:707–16.
Pastor V, Hirabayashi S, Karow A, Wehrle J, Kozyra EJ, Nienhold R, et al. Mutational landscape in children with myelodysplastic syndromes is distinct from adults: specific somatic drivers and novel germline variants. Leukemia. 2017;31:759–62.
West RR, Hsu AP, Holland SM, Cuellar-Rodriguez J, Hickstein DD. Acquired ASXL1 mutations are common in patients with inherited GATA2 mutations and correlate with myeloid transformation. Haematologica. 2014;99:276–81.
Stieglitz E, Liu YL, Emanuel PD, Castleberry RP, Cooper TM, Shannon KM, et al. Mutations in GATA2 are rare in juvenile myelomonocytic leukemia. Blood. 2014;123:1426–7.
Ganapathi KA, Townsley DM, Hsu AP, Arthur DC, Zerbe CS, Cuellar-Rodriguez J, et al. GATA2 deficiency-associated bone marrow disorder differs from idiopathic aplastic anemia. Blood. 2015;125:56–70.
Pasquet M, Bellanne-Chantelot C, Tavitian S, Prade N, Beaupain B, Larochelle O, et al. High frequency of GATA2 mutations in patients with mild chronic neutropenia evolving to MonoMac syndrome, myelodysplasia, and acute myeloid leukemia. Blood. 2013;121:822–9.
Gohring G, Michalova K, Beverloo HB, Betts D, Harbott J, Haas OA, et al. Complex karyotype newly defined: the strongest prognostic factor in advanced childhood myelodysplastic syndrome. Blood. 2010;116:3766–9.
Bodor C, Renneville A, Smith M, Charazac A, Iqbal S, Etancelin P, et al. Germ-line GATA2 p.THR354MET mutation in familial myelodysplastic syndrome with acquired monosomy 7 and ASXL1 mutation demonstrating rapid onset and poor survival. Haematologica. 2012;97:890–4.
Loyola VBP, Hirabayashi S, Pohl S, Kozyra EJ, Catala A, De Moerloose B, et al. Somatic genetic and epigenetic architecture of myelodysplastic syndromes arising from GATA2 deficiency. Blood. 2015;126(23):299 (abstract).
Kozyra EJ, Hirabayashi S, Loyola VBP, Przychodzen B, Karow A, Catala A, et al. Clonal mutational landscape of childhood myelodysplastic syndromes. Blood. 2015;126(23):1662 (abstract).
Inoue D, Kitaura J, Matsui H, Hou HA, Chou WC, Nagamachi A, et al. SETBP1 mutations drive leukemic transformation in ASXL1-mutated MDS. Leukemia. 2015;29:847–57.
Wang X, Muramatsu H, Okuno Y, Sakaguchi H, Yoshida K, Kawashima N, et al. GATA2 and secondary mutations in familial myelodysplastic syndromes and pediatric myeloid malignancies. Haematologica. 2015;100:e398–401.
Ding LW, Ikezoe T, Tan KT, Mori M, Mayakonda A, Chien W, et al. Mutational profiling of a MonoMAC syndrome family with GATA2 deficiency. Leukemia. 2017;31:244–5.
Acknowledgements
SH is supported by St. Luke's Life Science Institute Grant.
Author information
Authors and Affiliations
Contributions
SH and MWW wrote the paper, EK and CMN contributed with conception and figures.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
About this article

Cite this article
Hirabayashi, S., Wlodarski, M.W., Kozyra, E. et al. Heterogeneity of GATA2-related myeloid neoplasms. Int J Hematol 106, 175–182 (2017). https://doi.org/10.1007/s12185-017-2285-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-017-2285-2