
Abstract
Fanconi anemia (FA), the most common form of inherited bone marrow failure, predisposes to leukemia and solid tumors. FA is caused by the genetic disruption of a cellular pathway that repairs DNA interstrand crosslinks. The impaired function of this pathway, and the genetic instability that results, is considered the main pathogenic mechanism behind this disease. The identification of breast cancer susceptibility genes (for example, BRCA1/FANCS and BRCA2/FANCD1) as being major players in the FA pathway has led to a surge in molecular studies, resulting in the concept of the FA-BRCA pathway. In this review, we discuss recent advances in the molecular pathogenesis of FA from three viewpoints: (a) new FA genes, (b) modifier pathways that influence the cellular and clinical phenotypes of FA and (c) non-canonical functions of FA genes that may drive disease progression independently of deficient DNA repair. Potential therapeutic approaches for FA that are relevant to each will also be proposed.
Similar content being viewed by others

Introduction
Fanconi anemia (FA) is the most common inherited bone marrow failure syndrome. First described by the pediatrician Guido Fanconi [1], the clinical manifestations of this disease are a triad of aplastic anemia, predisposition to acute myelogenous leukemia (AML) and epithelial cancers, and congenital abnormalities. At the cellular level, FA is characterized by sensitivity to certain forms of DNA damage, most notably interstrand crosslinks (ICLs). This sensitivity is the basis for the diagnosis of FA, which involves a chromosome breakage assay in response to ICL-inducing agents such as mitomycin C (MMC) or diepoxybutane. The majority of FA patients exhibit abnormal physical findings. However, such findings may be absent or subtle, thereby presenting a diagnostic challenge.
Patients with FA are impaired in a pathway that repairs damaged DNA, and ICLs in particular. This defect is considered the major contributor to the disease state [2]. There are several reasons that support this statement. All FA genes identified to date have a demonstrable role in ICL repair. A DNA repair defect, which can be a cause of genetic instability, explains the cancer predisposition and ICL sensitivity seen in FA. Furthermore, experimental models have revealed that the bone marrow failure associated with the disease is caused by a response to genetic instability [3].
In this review, we discuss recent investigations into the molecular pathogenesis of FA within a framework comprised of three parts: genes, modifiers and drivers (Fig. 1). The discovery of new FA genes yields a more complete understanding of the canonical FA pathway, including insights into potentially targetable biochemistry and enzymology. By modifiers, we are referring to other metabolic and signaling pathways that influence cellular and clinical FA phenotypes. Finally, there is emerging evidence that certain FA genes have non-canonical functions outside of ICL repair that, when impaired, may also drive progression of the disease. For each part, opportunities with regard to the clinical management of FA are discussed.

A framework of how studies on the molecular pathogenesis of FA can improve understanding of disease progression. Identifying new FA genes leads to a more complete knowledge of those proteins involved in the canonical FA pathway. Modifier pathways contribute to FA by increasing the demands of DNA repair in cells that are deficient in the FA pathway. Drivers are DNA repair-independent (non-canonical) functions of FA genes that may contribute to disease progression. Potential therapeutic approaches that arise from the studies discussed in this review are bulleted
The FA-BRCA pathway
The 21 FA genes identified to date indicate that FA is caused by defects in a common pathway that repairs DNA ICLs. While basic studies have identified numerous proteins that are involved in ICL repair, the designation of an FA gene requires its association with actual clinical cases of FA. For example, although previous studies of BRCA1 lead one to predict that its disruption would cause FA, only the identification of FA patients with a loss of BRCA1 permitted its designation as FANCS [4, 5]. The clinical heterogeneity of FA, especially with regard to the development of bone marrow failure, has led to debate over what genes qualify for FA designation. For example, distinctions between FA genes and atypical FA genes, or FA-like genes, have been proposed [6, 7]. These will not be emphasized in this review, because there is insufficient data to establish that disruption of FA-like genes cause a clinically distinct disease.
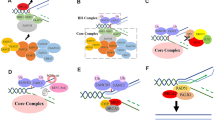
Typically, FA genes are categorized by their relationship to the monoubiquitination of two FA proteins, FANCD2 and FANCI, which form a heterodimer (the FANCI/D2 complex; Fig. 2). This grouping has both a molecular and a clinical basis. Eight FA genes encode proteins that comprise the multi-subunit ubiquitin ligase FA core complex that is responsible for the monoubiquitination reaction [8]. Because the majority of FA patients carry mutations in core complex proteins, FANCI/D2 monoubiquitination is impaired in greater than 90% of FA patients [9, 10]. FANCD2 monoubiquitination, in particular, is a critical modification that permits its recruitment to sites of DNA damage (a process known as foci formation) and the coordination of ICL repair by downstream FA proteins [11, 12]. It is therefore natural for FANCD2 monoubiquitination to have assumed a central position in the molecular biology of FA.

Schematic of the canonical FA pathway. FA genes can be organized by their relationship to FANCI/D2 monoubiquitination. The most recently identified FA genes (discussed in this review) are bolded in red. Because FANCV/REV7 has multiple activities, whether its polymerase function is the most relevant to FA is unclear
The FA pathway is activated in response to ICLs. The sensor protein that initially recognizes ICLs was recently identified as UHRF1 [13]. How this protein activates the FA pathway, beyond recruiting FANCD2 to ICLs, is not understood. At some point, activation of the pathway requires the phosphorylation of certain core complex proteins, as well as FANCI. The major kinase involved appears to be the Ataxia and Telangiectasia Rad3-related (ATR) kinase [14,15,16]. These phosphorylations promote the reciprocally dependent monoubiquitination of FANCI and FANCD2 by the core complex.
While FANCD2 monoubiquitination is essential for ICL repair, it is, by itself, a poor marker of an efficient FA pathway. In other words, having more monoubiquitinated FANCD2 within a cell does not necessarily improve, and in some cases inhibits, ICL repair. This is exemplified by studies demonstrating that USP1-mediated FANCD2 de-ubiquitination is required for both FANCD2 foci formation and ICL resistance [17, 18]. Furthermore, Usp1-deficient mice have an FA-like phenotype [19, 20]. Why de-ubiquitination of FANCD2 is important for ICL repair is unclear, although it might prevent the accumulation of this protein at genomic sites where it cannot function [21]. In addition, a recent study suggests that disrupting the proper equilibrium between monoubiquitinated and non-ubiquitinated FANCD2 by impairing USP1 leads to aberrantly low phosphorylation of FANCI, which may impede the function of the monoubiquitinated FANCI/D2 complex [22].
The FA proteins that act downstream of FANCI/D2 monoubiquitination are involved in various steps of the ICL repair process, ranging from unhooking of the ICL, lesion bypass with polymerase function, or homologous recombination (HR). Because mutations in such downstream genes are rare in FA, they tend to represent the more recently established players.
New genes
FANCR (RAD51)
The establishment of FANCR exemplifies the heterogeneous clinical presentation of FA. FANCR was identified in a patient who presented with microcephaly, slow growth, limb abnormalities and severe learning impairment [23]. Cells from the patient tested positive by chromosome breakage, were sensitive to multiple chemotherapeutic agents, and were intact in FANCD2 monoubiquitination, suggesting a mutation in a gene that acted downstream of this modification. The clinical presentation of this patient did not include bone marrow failure. For this reason, while meeting the criteria of FA by chromosome breakage, the patient was described as FA-like.
The causal mutation in this patient was a dominant-negative missense mutation in RAD51, a protein with a well established role in HR [24]. FANCR, therefore, represents the first autosomal dominant form of FA. The mutant RAD51 protein, RAD51A293T, was found to be impaired in DNA binding and D-loop formation, the latter being a biochemical marker of critical steps in HR. The ATPase activity of RAD51, which is essential for the HR function of the protein, was impaired in the RAD51A293T mutant.
In a separate study, a different dominant-negative RAD51 mutation (RAD51T131P) was identified in another FA patient [25]. Interestingly, this mutation disrupts ICL repair without affecting HR, suggesting an HR-independent role of RAD51 in the FA pathway.
FANCS (BRCA1)
The identification of BRCA1 as FANCS further establishes the concept of the FA-BRCA pathway that was first proposed after the identification of BRCA2 as FANCD1 [26]. FANCS was identified in a 23-year-old female with breast cancer, multiple congenital abnormalities and an FA-like presentation [4]. As with FANCR, bone marrow failure did not occur in this patient. The BRCA1 gene in this patient carried a missense mutation in the C terminal of one allele, and a frameshift mutation in exon 11 of the other.
This study was preceded by a report describing the biallelic loss of BRCA1 in a young ovarian cancer patient with multiple congenital abnormalities [5]. While the patient exhibited increased morbidity with cisplatin treatment, it appears that a diagnosis of FA was not fully established prior to her death. Thus, the identification of BRCA1 as an FA gene, which strictly speaking requires its association with at least two FA patients, is open to question.
Whether the development of breast or ovarian cancer will be a recurring feature of FANCS patients remains to be determined. Considering that BRCA1, BRCA2 and PALB2 have been established as FA genes, it is perhaps surprising that these cancers are not seen more often as presenting malignancies in FA. For example, reported malignancies in FANCD1/BRCA2 patients are leukemias, Wilm’s tumors and brain tumors [27, 28].
FANCT (UBE2T)
Most of the recently established FA genes function downstream of FANCI/D2 monoubiquitination. An exception is the ubiquitin-conjugating enzyme FANCT (UBE2T), which interacts with the ubiquitin-ligase component of the FA core complex, FANCL, and is required for FANCI/D2 monoubiquitination. Biallelic mutations in UBE2T were first reported to be causal for the clinical findings of three FA patients. Two of the patients were reported in a Japanese study [29]. Both of these patients exhibited bone marrow failure in the first decade of life, with one developing myelodysplastic syndrome (MDS) that progressed to AML. Whole exome sequencing of these patients did not reveal mutations in previously identified FA genes, but was able to establish that each of them carried a missense mutation in one UBE2T allele. One of the patients had a large deletion, while the other had a splice donor site mutation, in the other allele.
The third patient was reported in two separate studies [30, 31], and presented with multiple congenital abnormalities and a positive chromosome breakage assay. At the time of this review, the patient had not yet exhibited bone marrow failure. Malignancy was not reported in this patient. While whole exome sequencing of peripheral blood from the patient revealed a mutation in FANCA, western blot analysis showed normal FANCA levels. Furthermore, overexpression of wild-type FANCA did not rescue the impaired FANCD2 monoubiquitination that was observed in patient-derived fibroblasts. All other FA genes in the patient were normal by whole exome sequencing. Only RNA sequencing on patient-derived fibroblasts was able to identify decreased UBE2T RNA levels, which were subsequently found to result from compound heterozygous mutations in the UBE2T gene. This patient highlights the importance of analyzing different cell types (i.e., both fibroblasts and peripheral blood), as the patient exhibited somatic mosaicism, resulting in relatively normal UBE2T transcript levels and FA pathway function in the hematopoietic compartment. Thus, UBE2T would not have been identified as the causal gene in this patient if only peripheral blood was analyzed.
FANCU (XRCC2)
Mutations in XRCC2 were identified in a 2.5-year-old patient with congenital abnormalities that were suggestive of FA [32]. While the patient tested positive by chromosome breakage, peripheral blood counts were normal and aplastic anemia was absent. Whole exome sequencing of DNA from the patient, who was born to first cousin parents, identified germline mutations in several genes. However, complementation studies established that the RAD51 paralog XRCC2 was, in fact, the causal gene, thereby establishing it as FANCU [6]. XRCC2 acts downstream of FANCI/D2 monoubiquitination, and mediates HR by promoting RAD51 binding to double strand DNA breaks [33].
FANCV (REV7)
To this point, the most recently identified FA genes are, based on their molecular function, consistent with the importance of FANCD2 monoubiquitination (FANCT) and HR (FANCR, FANCS and FANCU) in ICL repair. When biallelic mutations in REV7, a subunit of the DNA polymerase ζ complex, were found to cause a classical presentation of FA (including aplastic anemia) in an eight-year-old female [34], the role of impaired translesion synthesis in the disease was suggested [35]. REV7 also plays a role in the choosing of non-homologous end joining (NHEJ) versus HR pathways. Specifically, it plays a role in suppressing HR, while promoting NHEJ [36, 37]. How losing this function contributes to the FA phenotype is not clear. Interestingly, REV7 is also involved in mitotic checkpoint regulation [38]. Studies implicating the FA pathway in this process are in their infancy [39]. The establishment of REV7 as FANCV, therefore, provides the impetus for further investigation into how the FA protein network may regulate mitosis.
Advances in whole exome sequencing have permitted the recent identification of several new FA genes. As patients with mutations in such genes are relatively rare when compared to other complementation groups (for example, FA-A), it is difficult to ascertain whether these patients will tend towards a distinct clinical course with regard to bone marrow failure and malignancy. As more FANCR to FANCV patients are identified, it is possible that the clinical spectrum of FA will broaden. This in turn may lead to the consideration of more refined clinical management strategies for different FA subtypes with regard to bone marrow failure and screening for malignancy. Such insights will require sufficient clinical data to permit reasonable conclusions.
Identification of the gene deficiencies in FA patients has obvious importance as treatment approaches involving gene therapy/editing are considered. Furthermore, as more players involved in the FA pathway, and their enzymology, are identified, opportunities to pharmacologically reactivate the pathway may arise. To date, drugs that can directly restore the function of the pathway in FA cells remain elusive. Given that greater than 90% of FA patients are unable to monoubiquitinate FANCD2, it makes sense to target this reaction for FA therapy. Yet, increasing FANCD2 monoubiquitination by inhibiting USP1, the de-ubiquitinating enzyme for FANCD2, is unlikely to restore the function of the FA pathway [17, 18]. Recent studies have focused on the basic biochemistry of the FANCD2 monoubiquitination reaction [40, 41]. In vitro studies such as these may provide critical information for the design of small molecules that can restore monoubiquitination in a manner that is therapeutically effective [42].
New modifiers
Until more opportunities to directly restore the FA pathway are revealed, other pathways that modify the cellular and clinical phenotypes of FA deserve attention. Understanding how such modifiers can be influenced by both environmental exposures and drug targeting may lead to clinical benefit for FA patients. Two modifier pathways that have been recently identified will be discussed here (Fig. 3).

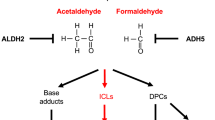
Modifier pathways that influence FA disease progression that are discussed in this review. Aldehydes act as a source of endogenous ICLs. In the context of an impaired FA pathway, overcoming the ability of aldehyde detoxifying enzymes to metabolize aldehydes results in the accumulation of ICLs that exacerbate disease progression. Hyperactive TGFβ signaling perturbs DNA repair mechanisms in a manner that further weakens ICL repair in FA cells. Drugs, or classes of drugs, that target specific components of the discussed modifier pathways are shown in red
Aldehyde metabolism and FA
Typically, while FA patients present with congenital and/or hematological findings at the time of their diagnosis, they usually do not have a history of exposure to exogenous ICL-inducing agents. If the pathogenesis of FA does indeed involve a defect in ICL repair, then an endogenous source of ICLs in FA patients needs to be established. It had been known for some time that aldehydes, which are byproducts of normal cellular metabolism, are capable of inducing ICLs [43]. Furthermore, in a chicken cell culture system, cells deficient in various FA genes were found to be sensitive to formaldehyde [44]. However, the first in vivo evidence suggesting a role of aldehydes in FA pathogenesis involved acetaldehyde, which is metabolized by aldehyde dehydrogenase 2 (ALDH2) to acetate. Mice deficient in both aldehyde dehydrogenase 2 and Fancd2 (Aldh2−/− Fancd2−/−) exhibited a more severe FA phenotype (with regard to aplastic anemia, leukemia and congenital defects) when compared to Fancd2−/− mice, which display a mild phenotype [45]. A similar study using Fanca−/− mice in place of Fancd2−/− mice reproduced several aspects of this study, suggesting that the interaction between impaired acetaldehyde metabolic and FA pathways may be generalizable to other FA genes [46].
An in vivo study involving formaldehyde soon followed. In addition to being a byproduct of numerous cellular de-methylation reactions, formaldehyde is present in a range of exogenous sources, hinting that lifestyle modifications may reduce exposure. Mice deficient in both the major enzyme for formaldehyde clearance, alcohol dehydrogenase 5, and Fancd2 (Adh5−/− Fancd2−/−) showed greater impairment in the bone marrow compartment, and increased dysfunction of the liver and kidneys, when compared to single knockout animals [47].
These studies suggest that the accumulation of endogenous aldehydes, induced by disrupting key enzymes in aldehyde metabolism, is sufficient to exacerbate at least some FA phenotypes. However, establishing ICLs as the pathogenic lesion resulting from excess aldehydes remains an elusive task, as both acetaldehyde and formaldehyde are capable of inducing a range of DNA lesions [43].
Remarkably, the findings from mouse studies were soon supported by studies of human subjects. In a study of Japanese FA patients [48], heterozygous presence of the ALDH2E504K allele, which suppresses ALDH2 activity in a dominant-negative manner and is present in approximately 50% of the Japanese population, was associated with a more rapid advancement to aplastic anemia. Progression to AML or MDS did not appear to be affected by the dominant-negative ALDH2. However, homozygosity of the ALDH2E504K allele (found in three of the FA patients in this study) was associated with very rapid progression to MDS and bone marrow failure. Whether the presence of the ALDH2E504K allele, in the context of FA, is associated with an increased risk of solid tumors remains to be determined.
The identification of aldehyde metabolism as a modifier of the FA phenotype has several clinical implications. First, identifying the ALDH2 genotype of FA patients, particularly those of East Asian descent, may be useful in determining their risk of developing bone marrow failure, and perhaps even malignancy. Second, as aldehydes exist in the environment, the importance of lifestyle modifications in the clinical management of FA patients has been increasingly recognized. Avoidance of alcohol and tobacco, for example, should be recommended to patients with FA. Third, the above-mentioned studies raise the possibility of using ALDH2 agonists in FA patients to delay the onset of bone marrow failure, and perhaps even malignancy [49]. One such agonist, Alda-1, was identified nearly a decade ago [50].
Pharmacologically modifying aldehyde metabolism for the treatment of FA might not require new drug development. In a recent study, the widely used diabetes drug, metformin, was found to protect an FA mouse model from chromosomal breakage and the development of malignancy [51]. The authors of this study found that this protective effect of metformin is potentially mediated through its activity as an aldehyde scavenger. This unexpected mechanism of action of metformin sets a precedent to screen existing drug libraries for aldehyde detoxifying/scavenging activity. The identification of such drugs, which may have established pharmacokinetic and safety data, could expedite therapeutic opportunities for FA patients.
TGFβ signaling and FA
The association of abnormal cytokine production with FA has been recognized for some time [52, 53]. Only recently was transforming growth factor beta (TGFβ) signaling specifically identified as being hyperactive in FA patients [54]. As with many studies involving the analysis of patient samples, it was difficult to infer from this study whether this increased TGFβ signaling contributed to the progression of FA.
This issue was resolved by a recent study [55] that initially used an shRNA library screen to identify genes that, when knocked down, could rescue the MMC sensitivity of a FANCA−/− fibroblast line. Genes involved in TGFβ signaling were among the top hits from this screen. This led to experiments involving the administration of a TGFβ neutralizing antibody to Fancd2−/− mice, the result being a rescue of hematopoietic stem cell function and prevention of bone marrow failure, via a mechanism that involves the upregulation of HR and the suppression of NHEJ. The involvement of TGFβ signaling in regulating DNA repair processes has been previously reported outside of the context of FA [56,57,58], as have the effects of suppressing NHEJ in FA cells [59]. The aforementioned study establishes that targeted inhibition of modifier pathways may help to circumvent the DNA repair defect in this disease.
Numerous drugs that target TGFβ signaling have been, and continue to be, developed, with ongoing clinical trials to establish their safety and efficacy, especially as oncology drugs [60]. Again, using such agents would be relatively expeditious, when compared to de novo drug design. In the setting of FA, such drugs may be clinically useful for several reasons. First, they may slow the progression of the disease with regard to bone marrow failure. Second, these inhibitors may improve the ex vivo expansion of stem cells derived from FA patients (which may have limited replicative potential [61]), thereby overcoming a barrier to gene therapy and gene editing approaches to treatment. Third, inhibitors to this pathway may offer new opportunities to manage the development of malignancy in FA patients. This final consideration is less straightforward. As typical DNA damaging chemotherapeutic agents (such as cisplatin) are of limited utility in the treatment of cancer in FA patients, targeting a signaling pathway is an appealing alternative. However, the biology of TGFβ with regard to cancer is pleiotropic, with it being a tumor suppressor early in cancer development, and a tumor promoter later on [62, 63]. The use of TGFβ in FA patients may, therefore, have unintended consequences with regard to cancer development and progression. While the bone marrow failure component of FA appears to be favorably affected by TGFβ inhibition, further studies of the interplay between TGFβ and FA pathways in carcinogenesis are needed.
New drivers
FA is a clinically heterogeneous disease. While environmental factors may play a role in this variability, it is possible that certain FA complementation groups are deficient in non-canonical functions that may drive certain aspects of disease progression. For example, while the predominant tumor suppressor function of an FA gene is mediated through its role in ICL repair, an additional function as a transcription factor may make FA patients who are deficient in that gene particularly susceptible to solid tumors, when compared to patients from other complementation groups. Another example would be the role of FANCV/REV7 in mitosis that was described above [38]. A discussion of autophagy as a potential driver of disease progression in FA follows (Fig. 4).

Alternate drivers of FA disease progression that are discussed in this review. The main driver of disease progression is a defect in ICL repair due to disruption of the canonical FA pathway. Impairment of a non-canonical function of an FA gene, such as autophagy, may drive the disease as well, through mechanisms that are independent of ICL repair
Autophagy and FA proteins
Cells have an intrinsic ability to remove unwanted structures by degradation. The discovery of this process, called autophagy, led to the award of the 2016 Nobel Prize in Physiology and Medicine. Mitophagy specifically refers to the autophagy of damaged mitochondria. Virophagy refers to the degradation of viruses. In a recent study [64], mice deficient in FANCC, and cells from FA-C patients, were found to be defective in both mitophagy and virophagy. This deficiency was found to be independent of the role of FANCC in DNA repair. In addition, knockdown of several other FA genes (FANCA, FANCF, FANCL, FANCD1, FANCD2 and FANCS) also impaired mitophagy, suggesting that this defect could contribute to disease progression in multiple complementation groups. These findings were, in part, validated in a separate study [65].
The implications of defective virophagy in FA include susceptibility to viral infections, and in particular those that may play a role in tumor development, such as Human Papilloma Virus (HPV). While the role of HPV in the development of cancer in FA patients is controversial [66, 67], the study discussed here provides a rationale for the already established guidelines of vaccinating FA patients against this virus.
Impaired mitophagy in FA patients could contribute to the generation of reactive oxygen species, resulting in both the activation of a cytokine-driven inflammatory state that contributes to bone marrow failure, and genomic damage that may further increase the risk of malignancy. The relationship between oxidative stress and FA was previously recognized [68], and the treatment of FA patients with antioxidants has been proposed [69]. Data showing a clear benefit of this are lacking, and concerns about toxicities associated with antioxidant use should be considered. Nevertheless, the role of FA proteins in mitophagy may prompt more controlled trials involving antioxidant use in FA patients. Similar statements can be made for modulators of cytokine signaling. Indeed, the mitophagy study corroborates a previous study reporting an abnormal inflammasome response in Fancc−/− mice [70].
While fascinating, the identification of non-canonical FA gene functions do not necessarily establish that they drive the progression of FA, even if they explain certain FA cellular phenotypes. Additional studies are needed before the contributions of impaired autophagy to FA can be fully determined.
Conclusions and future directions
In this review, we present a framework in which to consider how recent molecular studies on FA may yield new therapeutic opportunities and considerations. It is particularly satisfying that these recent discoveries are predicted by, and expand on, previous findings. The molecular biology of the FA pathway continues to bring forth fascinating insight into the mechanisms by which ICLs are repaired. As new FA genes are identified, gene therapy and editing approaches may offer therapeutic opportunities to complement or correct the relevant gene defect. From the standpoint of drug-based approaches, the molecular biology of the FA pathway has not yet yielded a clear therapeutic strategy to restore its function. However, our understanding is far from complete. Even the essential FANCD2 monoubiquitination reaction is only partially understood. Whether this process can be restored in a therapeutically meaningful way will require a more complete understanding of its biochemistry, and especially of the importance of de-ubiquitination.
The identification of modifier pathways and non-canonical functions of FA proteins, such as those discussed in this review, exemplifies the idea that therapeutic strategies may arise from indirect approaches. The ability to screen drug and genomic libraries in a high throughput manner may permit the identification of additional modifiers and drivers that could be modulated to slow progression of the disease.
FA is both a bone marrow failure syndrome and a cancer predisposition syndrome. Clinical studies that predate this review, as well as the molecular studies discussed here, have resulted in a myriad of therapeutic approaches to modify or correct the underlying cellular defect. However, these approaches have focused largely on treating the hematologic component of FA. This leaves the cancer predisposition, especially with regard to solid tumors that develop in FA patients. The obstacles to the clinical management of such cases are significant. As ICL-inducing agents are a mainstay of cancer chemotherapy, these drugs, and other DNA damaging modalities (including radiation therapy), are of limited utility in FA patients. At this time, reducing exposures (to tobacco and alcohol, for example), close monitoring and surgery feature heavily in the management of FA-related cancers. Emerging immune-based or targeted cancer therapies are currently being developed for non-FA-associated cancers [71], and may be applicable to FA-associated cancers. However, the efficacy of such approaches will depend on whether FA-associated cancers exhibit the appropriate biology for their use.
References
Lobitz S, Velleuer E. Guido Fanconi (1892–1979): a jack of all trades. Nat Rev Cancer. 2006;6:893–8.
Duxin JP, Walter JC. What is the DNA repair defect underlying Fanconi anemia? Curr Opin Cell Biol. 2015;26(37):49–60.
Ceccaldi R, Parmar K, Mouly E, Delord M, Kim JM, Regairaz M, et al. Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell. 2012;06(11):36–49.
Sawyer SL, Tian L, Kahkonen M, Schwartzentruber J, Kircher M, University of Washington Centre for Mendelian G, et al. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. 2015;5:135–42.
Domchek SM, Tang J, Stopfer J, Lilli DR, Hamel N, Tischkowitz M, et al. Biallelic deleterious BRCA1 mutations in a woman with early-onset ovarian cancer. Cancer Discov. 2013;3:399–405.
Park JY, Virts EL, Jankowska A, Wiek C, Othman M, Chakraborty SC, et al. Complementation of hypersensitivity to DNA interstrand crosslinking agents demonstrates that XRCC2 is a Fanconi anaemia gene. J Med Genet. 2016;53:672–80.
Bogliolo M, Surralles J. Fanconi anemia: a model disease for studies on human genetics and advanced therapeutics. Curr Opin Genet Dev. 2015;6(33):32–40.
Hodson C, Walden H. Towards a molecular understanding of the fanconi anemia core complex. Anemia. 2012;2012:926787.
Kee Y, D’Andrea AD. Molecular pathogenesis and clinical management of Fanconi anemia. J Clin Invest. 2012;122:3799–806.
Shimamura A, Montes de Oca R, Svenson JL, Haining N, Moreau LA, Nathan DG, et al. A novel diagnostic screen for defects in the Fanconi anemia pathway. Blood. 2002;15(100):4649–54.
Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, et al. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001;7:249–62.
Taniguchi T, Garcia-Higuera I, Xu B, Andreassen PR, Gregory RC, Kim ST, et al. Convergence of the fanconi anemia and ataxia telangiectasia signaling pathways. Cell. 2002;17(109):459–72.
Liang CC, Zhan B, Yoshikawa Y, Haas W, Gygi SP, Cohn MA. UHRF1 is a sensor for DNA interstrand crosslinks and recruits FANCD2 to initiate the Fanconi anemia pathway. Cell Rep. 2015;31(10):1947–56.
Ishiai M, Kitao H, Smogorzewska A, Tomida J, Kinomura A, Uchida E, et al. FANCI phosphorylation functions as a molecular switch to turn on the Fanconi anemia pathway. Nat Struct Mol Biol. 2008;15:1138–46.
Collins NB, Wilson JB, Bush T, Thomashevski A, Roberts KJ, Jones NJ, et al. ATR-dependent phosphorylation of FANCA on serine 1449 after DNA damage is important for FA pathway function. Blood. 2009;5(113):2181–90.
Singh TR, Ali AM, Paramasivam M, Pradhan A, Wahengbam K, Seidman MM, et al. ATR-dependent phosphorylation of FANCM at serine 1045 is essential for FANCM functions. Cancer Res. 2013;15(73):4300–10.
Oestergaard VH, Langevin F, Kuiken HJ, Pace P, Niedzwiedz W, Simpson LJ, et al. Deubiquitination of FANCD2 is required for DNA crosslink repair. Mol Cell. 2007;14(28):798–809.
Liang Q, Dexheimer TS, Zhang P, Rosenthal AS, Villamil MA, You C, et al. A selective USP1-UAF1 inhibitor links deubiquitination to DNA damage responses. Nat Chem Biol. 2014;10:298–304.
Kim JM, Parmar K, Huang M, Weinstock DM, Ruit CA, Kutok JL, et al. Inactivation of murine Usp1 results in genomic instability and a Fanconi anemia phenotype. Dev Cell. 2009;16:314–20.
Parmar K, Kim J, Sykes SM, Shimamura A, Stuckert P, Zhu K, et al. Hematopoietic stem cell defects in mice with deficiency of Fancd2 or Usp1. Stem Cells. 2010;28:1186–95.
Ogrunc M, Martinez-Zamudio RI, Sadoun PB, Dore G, Schwerer H, Pasero P, et al. USP1 regulates cellular senescence by controlling genomic integrity. Cell Rep. 2016;17(15):1401–11.
Cheung RS, Castella M, Abeyta A, Gafken PR, Tucker N, Taniguchi T. Ubiquitination-linked phosphorylation of the FANCI ST/Q cluster contributes to activation of the FANCI/D2 complex. Cell Rep. 2017. doi:10.1016/j.celrep.2017.05.081.
Ameziane N, May P, Haitjema A, van de Vrugt HJ, van Rossum-Fikkert SE, Ristic D, et al. A novel Fanconi anaemia subtype associated with a dominant-negative mutation in RAD51. Nat Commun. 2015;18(6):8829.
Prakash R, Zhang Y, Feng W, Jasin M. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol. 2015;01(7):a016600.
Wang AT, Kim T, Wagner JE, Conti BA, Lach FP, Huang AL, et al. A dominant mutation in human RAD51 reveals its function in DNA interstrand crosslink repair independent of homologous recombination. Mol Cell. 2015;06(59):478–90.
Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;26(297):606–9.
Wagner JE, Tolar J, Levran O, Scholl T, Deffenbaugh A, Satagopan J, et al. Germline mutations in BRCA2: shared genetic susceptibility to breast cancer, early onset leukemia, and Fanconi anemia. Blood. 2004;15(103):3226–9.
Reid S, Renwick A, Seal S, Baskcomb L, Barfoot R, Jayatilake H, et al. Biallelic BRCA2 mutations are associated with multiple malignancies in childhood including familial Wilms tumour. J Med Genet. 2005;42:147–51.
Hira A, Yoshida K, Sato K, Okuno Y, Shiraishi Y, Chiba K, et al. Mutations in the gene encoding the E2 conjugating enzyme UBE2T cause Fanconi anemia. Am J Hum Genet. 2015;04(96):1001–7.
Rickman KA, Lach FP, Abhyankar A, Donovan FX, Sanborn EM, Kennedy JA, et al. Deficiency of UBE2T, the E2 ubiquitin ligase necessary for FANCD2 and FANCI ubiquitination, causes FA-T subtype of fanconi anemia. Cell Rep. 2015;7(12):35–41.
Virts EL, Jankowska A, Mackay C, Glaas MF, Wiek C, Kelich SL, et al. AluY-mediated germline deletion, duplication and somatic stem cell reversion in UBE2T defines a new subtype of Fanconi anemia. Hum Mol Genet. 2015;15(24):5093–108.
Shamseldin HE, Elfaki M, Alkuraya FS. Exome sequencing reveals a novel Fanconi group defined by XRCC2 mutation. J Med Genet. 2012;49:184–6.
Suwaki N, Klare K, Tarsounas M. RAD51 paralogs: roles in DNA damage signalling, recombinational repair and tumorigenesis. Semin Cell Dev Biol. 2011;22:898–905.
Bluteau D, Masliah-Planchon J, Clairmont C, Rousseau A, Ceccaldi R, Dubois d’Enghien C, et al. Biallelic inactivation of REV7 is associated with Fanconi anemia. J Clin Invest. 2016;01(126):3580–4.
Lee YS, Gregory MT, Yang W. Human Pol zeta purified with accessory subunits is active in translesion DNA synthesis and complements Pol eta in cisplatin bypass. Proc Natl Acad Sci USA. 2014;25(111):2954–9.
Boersma V, Moatti N, Segura-Bayona S, Peuscher MH, van der Torre J, Wevers BA, et al. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5′ end resection. Nature. 2015;28(521):537–40.
Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature. 2015;28(521):541–4.
Listovsky T, Sale JE. Sequestration of CDH1 by MAD2L2 prevents premature APC/C activation prior to anaphase onset. J Cell Biol. 2013;14(203):87–100.
Nalepa G, Enzor R, Sun Z, Marchal C, Park SJ, Yang Y, et al. Fanconi anemia signaling network regulates the spindle assembly checkpoint. J Clin Invest. 2013;3(123):3839–47.
Swuec P, Renault L, Borg A, Shah F, Murphy VJ, van Twest S, et al. The FA core complex contains a homo-dimeric catalytic module for the symmetric mono-ubiquitination of FANCI-FANCD2. Cell Rep. 2017;18:611–23.
van Twest S, Murphy VJ, Hodson C, Tan W, Swuec P, O’Rourke JJ, et al. Mechanism of ubiquitination and deubiquitination in the Fanconi Anemia pathway. Mol Cell. 2016;65(2):247–59.
Tan W, Deans AJ. Perspective: a defined role for multiple Fanconi anemia gene products in DNA-damage-associated ubiquitination. Exp Hematol. 2017 Mar 16.
Voulgaridou GP, Anestopoulos I, Franco R, Panayiotidis MI, Pappa A. DNA damage induced by endogenous aldehydes: current state of knowledge. Mutat Res. 2011;03(711):13–27.
Ridpath JR, Nakamura A, Tano K, Luke AM, Sonoda E, Arakawa H, et al. Cells deficient in the FANC/BRCA pathway are hypersensitive to plasma levels of formaldehyde. Cancer Res. 2007;1(67):11117–22.
Langevin F, Crossan GP, Rosado IV, Arends MJ, Patel KJ. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature. 2011;7(475):53–8.
Oberbeck N, Langevin F, King G, de Wind N, Crossan GP, Patel KJ. Maternal aldehyde elimination during pregnancy preserves the fetal genome. Mol Cell. 2014;18(55):807–17.
Pontel LB, Rosado IV, Burgos-Barragan G, Garaycoechea JI, Yu R, Arends MJ, et al. Endogenous Formaldehyde Is a Hematopoietic Stem Cell Genotoxin and Metabolic Carcinogen. Mol Cell. 2015;01(60):177–88.
Hira A, Yabe H, Yoshida K, Okuno Y, Shiraishi Y, Chiba K, et al. Variant ALDH2 is associated with accelerated progression of bone marrow failure in Japanese Fanconi anemia patients. Blood. 2013;31(122):3206–9.
Chen CH, Ferreira JC, Gross ER, Mochly-Rosen D. Targeting aldehyde dehydrogenase 2: new therapeutic opportunities. Physiol Rev. 2014;94:1–34.
Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;12(321):1493–5.
Zhang QS, Tang W, Deater M, Phan N, Marcogliese AN, Li H, et al. Metformin improves defective hematopoiesis and delays tumor formation in Fanconi anemia mice. Blood. 2016;15(128):2774–84.
Rosselli F, Sanceau J, Wietzerbin J, Moustacchi E. Abnormal lymphokine production: a novel feature of the genetic disease Fanconi anemia. I. Involvement of interleukin-6. Hum Genet. 1992;89:42–8.
Dufour C, Corcione A, Svahn J, Haupt R, Poggi V, Beka’ssy AN, et al. TNF-alpha and IFN-gamma are overexpressed in the bone marrow of Fanconi anemia patients and TNF-alpha suppresses erythropoiesis in vitro. Blood. 2003;15(102):2053–9.
Korthof ET, Svahn J, Peffault de Latour R, Terranova P, Moins-Teisserenc H, Socie G, et al. Immunological profile of Fanconi anemia: a multicentric retrospective analysis of 61 patients. Am J Hematol. 2013;88:472–6.
Zhang H, Kozono DE, O’Connor KW, Vidal-Cardenas S, Rousseau A, Hamilton A, et al. TGF-beta Inhibition rescues hematopoietic stem cell defects and bone marrow failure in Fanconi anemia. Cell Stem Cell. 2016;05(18):668–81.
Liu L, Zhou W, Cheng CT, Ren X, Somlo G, Fong MY, et al. TGFbeta induces “BRCAness” and sensitivity to PARP inhibition in breast cancer by regulating DNA-repair genes. Mol Cancer Res. 2014;12:1597–609.
Kim MR, Lee J, An YS, Jin YB, Park IC, Chung E, et al. TGFbeta1 protects cells from gamma-IR by enhancing the activity of the NHEJ repair pathway. Mol Cancer Res. 2015;13:319–29.
Pal D, Pertot A, Shirole NH, Yao Z, Anaparthy N, Garvin T, et al. TGF-beta reduces DNA ds-break repair mechanisms to heighten genetic diversity and adaptability of CD44 +/CD24- cancer cells. Elife. 2017;16:6.
Adamo A, Collis SJ, Adelman CA, Silva N, Horejsi Z, Ward JD, et al. Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol Cell. 2010;9(39):25–35.
Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov. 2012;11:790–811.
Habi O, Delisle MC, Messier N, Carreau M. Lack of self-renewal capacity in Fancc−/− stem cells after ex vivo expansion. Stem Cells. 2005;23:1135–41.
Wakefield LM, Roberts AB. TGF-beta signaling: positive and negative effects on tumorigenesis. Curr Opin Genet Dev. 2002;12:22–9.
Roberts AB, Wakefield LM. The two faces of transforming growth factor beta in carcinogenesis. Proc Natl Acad Sci USA. 2003;22(100):8621–3.
Sumpter R Jr, Sirasanagandla S, Fernandez AF, Wei Y, Dong X, Franco L, et al. Fanconi anemia proteins function in mitophagy and immunity. Cell. 2016;05(165):867–81.
Shyamsunder P, Esner M, Barvalia M, Wu YJ, Loja T, Boon HB, et al. Impaired mitophagy in Fanconi anemia is dependent on mitochondrial fission. Oncotarget. 2016;06(7):58065–74.
Alter BP, Giri N, Savage SA, Quint WG, de Koning MN, Schiffman M. Squamous cell carcinomas in patients with Fanconi anemia and dyskeratosis congenita: a search for human papillomavirus. Int J Cancer. 2013;15(133):1513–5.
Sauter SL, Wells SI, Zhang X, Hoskins EE, Davies SM, Myers KC, et al. Oral human papillomavirus is common in individuals with Fanconi anemia. Cancer Epidemiol Biomarkers Prev. 2015;24:864–72.
Du W, Adam Z, Rani R, Zhang X, Pang Q. Oxidative stress in Fanconi anemia hematopoiesis and disease progression. Antioxid Redox Signal. 2008;10:1909–21.
Zhang QS, Marquez-Loza L, Eaton L, Duncan AW, Goldman DC, Anur P, et al. Fancd2−/− mice have hematopoietic defects that can be partially corrected by resveratrol. Blood. 2010;9(116):5140–8.
Garbati MR, Hays LE, Keeble W, Yates JE, Rathbun RK, Bagby GC. FANCA and FANCC modulate TLR and p38 MAPK-dependent expression of IL-1beta in macrophages. Blood. 2013;31(122):3197–205.
Moreira J, Tobias A, O’Brien MP, Agulnik M. Targeted therapy in head and neck cancer: an update on current clinical developments in epidermal growth factor receptor-targeted therapy and immunotherapies. Drugs. 2017;77:843–57.
Acknowledgements
This work was supported by Howard Hughes Medical Institute, Fanconi Anemia Research Fund (to T.T.), the NIH/NCI R01 CA125636 (to T.T.) and JSPS KAKENHI15K21771 (to T.T.). R.S.C. was supported by the NIH/Ruth L. Kirschstein National Research Service Award T32 CA009351 and by the NIH/NICHD University of Washington Child Health Research Center K12 HD043376.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
About this article

Cite this article
Cheung, R.S., Taniguchi, T. Recent insights into the molecular basis of Fanconi anemia: genes, modifiers, and drivers. Int J Hematol 106, 335–344 (2017). https://doi.org/10.1007/s12185-017-2283-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-017-2283-4