
Abstract
Steady-state hematopoiesis is maintained by slowly dividing, self-renewing hematopoietic stem cells (HSCs) and their offspring, lineage-specified downstream progenitors in bone marrow (BM). It was previously thought that hematopoietic stresses such as infection or other inflammatory stimuli, are mostly recognized by terminally differentiated immune cells, i.e., front-line defenders at the local site of reaction, and that they produce factors that directly act on hematopoietic stem and progenitors (HSPCs) in BM and subsequently stimulate them to rebuild and sustain the hemato-lymphatic system. However, accumulating evidence now indicates that primitive HSPCs, as well as microenvironmental cells in BM are also able to sense systemically migrating hematopoietic stress signals, and respond by orchestrating on-site hematopoiesis via direct and indirect mechanisms. While inflammation has many beneficial roles in activating the immune system for defense or facilitating tissue repair, it also shows detrimental effects if sustained chronically, i.e., might lead to HSPC damage as bone marrow failure or leukemia. Thus, inflammation requires tight control of initiation and termination in time and space dependent manner.
Similar content being viewed by others
Steady state hematopoiesis
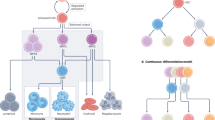
Millions of blood cells are continuously being born and are dying every day throughout the lifetime of an individual [1]. High throughput cell production and robust regenerative capacity of hematopoiesis is sustained by a rare population of hematopoietic stem cells (HSCs) which are preserved in bone marrow (BM). HSCs can give rise to themselves, i.e., self-renew, and at the same time, differentiate to multipotent progenitors (MPPs) with limited self-renewal, but high proliferative capacity, [2,3,4,5]. Subsequently, MPPs generate lineage committed downstream progenitors, such as common myeloid (CMP) or common lymphoid progenitors (CLP) that are specified to myeloid and lymphoid lineage, respectively. The stepwise hematopoietic lineage commitment is tightly regulated by both cell-intrinsic (transcription, epigenetics, metabolism, etc.) [6] and cell-extrinsic mechanisms (fluid factors, cell–cell interaction, etc.). A recent single cell analysis study has identified intermediate myeloid progenitor populations that show hierarchical, metastable genomic and regulatory states [7], although another study did not find such a cell population with mixed transcriptional priming [8]. In contrast, it has been shown that cell-extrinsic signals, e.g., cytokines and chemokines, can instruct cell lineage specification, as demonstrated by the case of myeloid supporting cytokines [9], and could activate lineage specific factors, e.g., PU.1 or GATA1, that execute and reinforce differentiation [10].
Since steady-state hematopoiesis occurs dominantly in adult mammalian BM, it had been a long-standing hypothesis that BM microenvironment contains specialized cells that provide HSC and progenitors (HSPCs) with factors necessary and essential for their maintenance, survival and differentiation [11]. Along with development of advanced microscopy-based imaging, and temporally and spatially controlled genetic modification of animals, the cellular and molecular components of BM environment, the so-called “niche”, has been uncovered for the last decades (reviewed in [12, 13]): there are major regions of BM that have been shown to have niche functions to support HSC homeostasis, the so-called endosteal and perivascular niche. The endosteal niche is where HSCs are in close contact with bone-lining osteoblastic cells. Despite of the fact that osteoblastic cells have been first demonstrated as the key HSC niche component [14, 15], several evidences subsequently raised concerns that the HSC function might not be regulated directly by osteoblastic niche but rather indirectly via fluid factors produced by the adjacent osteoblasts. On the other hand, the perivascular niche is composed of mesenchymal stromal cells (MSCs) and endothelial cells (ECs) that secrete HSC maintenance factors, such as stem cell factor (SCF) [16] and CXCL12 [17]. Although MSCs are likely a heterogeneous population, consisting of one surrounding the vessels and another forming a reticular network throughout BM, it is clear that they play critical roles for HSC homeostasis. However, the cellular heterogeneity and hierarchy within MSCs and ECs, and the functional relationship between distinct populations remains to be better defined.
Inflammation-induced activation of early hematopoiesis in bone marrow
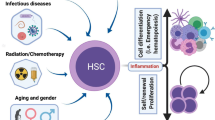
In steady state, most HSCs are kept in quiescence, i.e., in G0 of cell cycle, and are activated only infrequently to divide about once every months [12, 18], which is in stark contrast to actively proliferating progenitors. Recent studies have demonstrated that HSCs rarely contribute to hematopoiesis [19, 20], while other studies have claimed that they are continuously engaged in blood formation [21]. However, when the pool of short-lived mature hematopoietic cells is consumed, and therefore, needs to be replenished, e.g., in the case of naturally occurring hematopoietic stresses such as life-threatening blood loss, infection or other inflammation, many HSCs can be activated to divide and generate mature lineage cells that are lost. HSC activation is mediated by sensing the demand of hematopoietic cell production through extracellularly expressed surface receptors such as cytokine receptors, chemokine receptors, or pattern recognition receptors (PRRs), and the activation of respective receptors and their subsequent intracellular signaling lead to migration, proliferation and/or differentiation. Understanding how hematopoiesis is altered upon hematopoietic stress has become a major focus of research in the field, and has been addressed using different animal models challenged with hematopoietic stresses, such as infection or chronic inflammation (Fig. 1).

Hematopoietic lineage commitment from hematopoietic stem cells to mature cells. Hematopoietic stem cells (HSCs) at the apex of hematopoietic hierarchy self-renew (circle arrow) and give rise to multipotent progenitors (MPP) with limited self-renewal potential (dashed circle arrow). MPP produce myeloid and lymphoid progenitor cells that further differentiate to myeloid and lymphoid cells, respectively. This lineage commitment process is regulated by both cell-extrinsic factors, e.g., fluid factors, adhesion molecules, and cell-intrinsic factors, e.g., transcription factors, epigenetic modification, metabolism
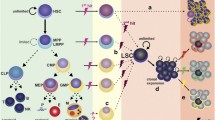
One of the hematopoietic stresses that are highly conserved in evolution and relevant to clinics is infection. In response to viral or bacterial infection, professional immune cells in the front line of defense becomes rapidly activated for terminal differentiation and functional maturation, and exert their specialized immune function to fight against invading pathogens. Since immune cells are short-lived and eventually eliminated after fulfilling their function, early hematopoiesis needs to detect the danger signals from the site of infection, via respective receptors, and translate them to direct hematopoietic output to rebuild and sustain the immune system for future hematopoietic challenges. Recent studies have proposed three possible, not mutually exclusive mechanisms (Fig. 2) [22]: (a) a cytokine-dependent mechanism, in which immune cells produce an array of pro-inflammatory cytokines/chemokines including type I Interferons (IFNs) at the local site of infection and/or in the BM, and act on respective receptors to activate the downstream signaling and induce proliferation/differentiation in HSPCs (Fig. 2➀, ➁) [23,24,25,27]; (b) a pathogen sensing receptor-dependent mechanism, where pathogen-associated molecular patterns (PAMPs) are recognized by HSPCs themselves via the respective pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs), Nod-like receptor (NLRs), and enhance their proliferation, differentiation and migration (Fig. 2➂) [28,29,30,31,32]; (c) a mechanism that combines both of the above. Direct TLR activation in HSPCs results in production of pro-inflammatory cytokines such as IL-6, which cell intrinsically, either in a paracrine or autocrine manner, enhance proliferation and myeloid differentiation under neutropenic conditions in vivo (Fig. 2➃) [33].

Direct and indirect infection-induced hematopoietic activation in BM. Viral infection is primarily recognized by immune cells such as cytotoxic T lymphocytes (CTL), macrophages that upon activation secrete interferons (IFNs) or granulocyte colony-stimulating factor (G-CSF) (➀). IFNs activate HSPCs to proliferation and reduce their self-renewal (➁). Pathogen-associated molecule patterns (PAMPs) derived from bacterial infection directly act on pattern recognition receptors (PRRs) expressed on hematopoietic stem and progenitor cells (HSPCs) and direct their differentiation to myelopoiesis (➂). At the same time, HSPCs are induced to secrete pro-inflammatory cytokines such as interleukin-6 (IL-6) that activate themselves in a paracrine or autocrine manner (➃). IFNs produced by immune cells upon infection also triggers production of IL-6 and nitric-oxide synthase (NOS) from mesenchymal stromal cells (MSCs) that modulate immune cell differentiation and function (➄), while G-CSF secreted by immune cells suppresses CXCL12 and IL-6 secretion in MSCs resulting in the reduction of B lymphopoiesis (➅). PRR activation on MSCs leads to the production of CCL2 that induces monocyte trafficking (➆). Lastly, G-CSF or granulocyte macrophage colony-stimulating factor (GM-CSF) are produced by endothelial cells in response to PRR activation and enhance myeloid differentiate of HSPCs (➇)
These findings demonstrate HSPC response to infection by which non-immune HSPCs are able to sense systemically spreading infection via indirect or direct mechanism and adapt hematopoiesis during a course of infection. However, if inflammation does not cease timely and is sustained for longer times, it might ultimately cause detrimental effects, such as continuous tissue damage/repair, chronic diseases, and cancer. It has been recently shown that sustained IFN-α/γ activation during chronic infection impairs HSC function, and can ultimately lead to BM failure [34, 35]. IL-1 directly activates proliferation and differentiates HSCs, resulting in their reduced self-renewal potential [36]. Chronic TNF-α signaling is associated with myelodysplastic syndrome and bone marrow failure [37]. Therefore, it is critical that inflammatory responses stop when no longer needed.
Inflammation-induced activation of the hematopoietic niche
Many lines of evidences have revealed that BM niche cells including MSCs and ECs express an array of cytokine/chemokine receptors and PRRs, and recognize an inflammatory milieu that is generated locally or systemically upon hematopoietic challenges. The activation of inflammation-sensing receptors in MSCs and ECs modulates their cellular functions, and triggers production of a second wave of other inflammatory factors that influence hematopoiesis: pro-inflammatory cytokines regulate cytokine/chemokine expression profile in MSCs. IFNγ alone or in combination with tumor necrosis factor (TNF) or IL-1 induces the production of nitric-oxide synthase (NOS) or prostaglandin E (PGE)-2 in MSCs, thereby inhibiting T cell or NK cell activation, respectively (Fig. 2➄) [38]. IL-6 secreted by MSCs polarize macrophages into anti-inflammatory phenotype that contribute to insulin resistance in type 2 diabetes [39]. Granulocyte colony-stimulating factor (G-CSF), a myeloid-supporting cytokine, up-regulated upon infection, directs MSC to enhance myelopoiesis by suppressing B lymphopoiesis (Fig. 2➅) [40]. G-CSF also suppresses production of CXCL12 from MSCs in BM and mobilizes HSC into circulating blood [41]. Ligation of TLRs expressed on MSCs leads to modulation of their differentiation, proliferation, migration, immunomodulation and bone regenerating potential [42,43,44]. Simultaneously, upon TLR stimulation, MSCs induce monocyte egress from BM by expressing monocyte recruiting chemokine, CCL2 (MCP1) (Fig. 2➆) [45, 46].
In addition to ECs in the peripheral organs often exposed to infection, BM-resident ECs express multiple PRRs, and contribute to regulation of various immune responses against systemic infection [26]. Several studies have shown that G-CSF is predominantly produced from ECs in BM in response to TLR4 activation, and drives emergency granulopoiesis, represented by rapid neutrophil production in BM [47, 48], and neutrophil recruitment to the site of infection [49]. This is in part in line with the previous finding that upon stimulation with IL-1β and TNF-α, BM-derived ECs produce granulocyte macrophage colony-stimulating factor (GM-CSF) which recruits neutrophils and expands HPCs in the BM (Fig. 2➇) [50, 51]. Pro-inflammatory cytokines regulate hematopoiesis not only via cytokine secreted from ECs in BM but also via cell–cell adhesion molecule expressed on ECs. In vivo administration of TNF-α and LPS expands the HPC population through augmentation of Notch signals between HPC and ECs in the BM [52]. Besides MSC- or EC-dependent hematopoietic modulation, it is of note that through MSC–EC interaction, MSCs up-regulates IL-6 production and modulates the responsiveness of ECs to inflammatory cytokines [53].
Altogether, these studies suggest that inflammation directly affects cellular functions of both MSCs and ECs, and regulates hematopoiesis mainly through modulation of their cytokine profiles.
Inflammation and hematopoietic malignancies
Inflammation has been long considered as one of the risk factors for cancer development in many tissues, especially in solid organs [54]. However, there has not been any strong evidence supporting a potential role of inflammation as a driving force for development of hematologic malignancies. Recent development of high resolution sequencing has shown that malignant clones in some hematologic malignancies accumulate somatic mutations in inflammatory genes including NFκB, myeloid differentiation primary response gene 88, TLR4, and TNF receptor-associated factors, and thereby, are hyper-proliferative or resistant to apoptosis [55]. Other evidence to support inflammation-driven leukemogenesis are shown by the population-based epidemiological studies in the Swedish cancer registry which analyzed the database of patients with hematologic malignancies arising from HSC or myeloid progenitors, and showed significantly strong association between history of infection or autoimmunity, and the high risk of incidence of hematologic malignancies [56, 57]. It has been recently shown that dysfunction of BM niche also affects blood homeostasis, and in some cases although rare events, contribute towards initiation of hematologic malignancies, suggesting a leukemic niche [58]. Specific genetic deletion of Shwachman–Diamond Syndrome (SDS) in MSCs results in myelodysplasia in mice, some of which develop secondary leukemia [59]. Furthermore, MSC induces genotoxic stress in HSC via danger-associated molecule patterns via a TLR4 signaling axis, and the same mechanism was found in human SDS patients, suggesting mesenchymal inflammatory cues that induce genetic ablation in HSC and predicts disease outcome in human SDS [60].
Taken together, these findings have suggested that inflammation fosters accumulation of genetic alterations, possibly by preventing pre-malignant HSPC clones from apoptosis and enhancing more mutations, and drive malignant transformation to initiation and progression of myeloid malignancies [61]. However, mechanistic insight on how chronically sustained inflammatory stimuli promote acquisition of genetic events in pre-leukemic clones, and contribute to clonal evolution in the development of hematologic malignancies, and to what extend cell-intrinsic and -extrinsic inflammation is relevant, remains to be determined at the cellular and molecular level.
Future direction
Recent studies have advanced our understanding of how early hematopoiesis respond to inflammation, and direct hematopoiesis via indirect (inflammatory mediators) or direct mechanisms (pathogen/danger sensors) to cope with exogenous and endogenous hematopoietic challenges. Depending on the degree or type of inflammation, the hematopoietic hierarchy might be organized differently from the one in steady state, as indicated by a recent study that identified a new cell population, emerging only upon inflammation: stem cell-like megakaryocyte-committed progenitors exist in a dormant status and thereby contribute little to megakaryopoiesis in the steady state. However, in response to acute inflammation, they become metabolically active and rapidly produce platelets for replenishment [62]. Since inflammation alters the immune-phenotype of hematopoietic cells, it is difficult to identify each cell population at different hematopoietic hierarchy simply by phenotype. Thus, although challenging, it would be important to determine how hematopoiesis is affected by inflammation and what are the commonalities and differences in various inflammatory situations at the cellular and molecular level, and how hematopoietic cell-intrinsic and -extrinsic signals contribute to inflammation-adapted hematopoiesis, respectively. Furthermore, there might be common biological changes in hematopoiesis in inflammation and aging, proposing inflamm-aging [63]. Given that low grade of inflammation accelerates aging and possibly contribute to aging-associated leukemogenesis, eliminating unwanted inflammatory responses will be a potential approach to preserve the beneficial role of inflammation in regulation of hematopoiesis and immunity while preventing a functional decline in hematopoiesis and the emergence of malignant clones.
References
Fliedner TM, Graessle D, Paulsen C, Reimers K. Structure and function of bone marrow hemopoiesis: mechanisms of response to ionizing radiation exposure. Cancer Biother Radiopharm. 2002;17:405–26.
Morrison SJ, Wandycz AM, Hemmati HD, Wright DE, Weissman IL. Identification of a lineage of multipotent hematopoietic progenitors. Development. 1997;124:1929–39.
Pietras EM, Reynaud D, Kang Y-A, Carlin D, Calero-Nieto FJ, Leavitt AD, et al. Functionally distinct subsets of lineage-biased multipotent progenitors control blood production in normal and regenerative conditions. Cell Stem Cell. 2015;17:35–46.
Laslo P, Spooner CJ, Warmflash A, Lancki DW, Lee H-J, Sciammas R, et al. Multilineage transcriptional priming and determination of alternate hematopoietic cell fates. Cell. 2006;126:755–66.
Adolfsson J, Borge OJ, Bryder D, Theilgaard-Mönch K, Astrand-Grundström I, Sitnicka E, et al. Upregulation of Flt3 expression within the bone marrow Lin(−)Sca1(+)c-kit(+) stem cell compartment is accompanied by loss of self-renewal capacity. Immunity. 2001;15:659–69.
Cabezas-Wallscheid N, Klimmeck D, Hansson J, Lipka DB, Reyes A, Wang Q, et al. Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell. 2014;15:507–22.
Olsson A, Venkatasubramanian M, Chaudhri VK, Aronow BJ, Salomonis N, Singh H, et al. Single-cell analysis of mixed-lineage states leading to a binary cell fate choice. Nature. 2016;537:698–702.
Paul F, Arkin Y, Giladi A, Jaitin DA, Kenigsberg E, Keren-Shaul H, et al. Transcriptional heterogeneity and lineage commitment in myeloid progenitors. Cell. 2015;163:1663–77.
Rieger MA, Hoppe PS, Smejkal BM, Eitelhuber AC, Schroeder T. Hematopoietic cytokines can instruct lineage choice. Science (New York, N.Y.). 2009;325:217–8.
Hoppe PS, Schwarzfischer M, Loeffler D, Kokkaliaris KD, Hilsenbeck O, Moritz N, et al. Early myeloid lineage choice is not initiated by random PU.1 to GATA1 protein ratios. Nature. 2016;535:299–302.
Schofield R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells. 1978;4:7–25.
Nakamura-Ishizu A, Takizawa H, Suda T. The analysis, roles and regulation of quiescence in hematopoietic stem cells. Development. 2014;141:4656–66.
Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505:327–34.
Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–6.
Zhang J, Niu C, Ye L, Huang H, He X, Tong W-G, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–41.
Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. 2013;495:231–5.
Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977–88.
Takizawa H, Manz MG. In vivo divisional tracking of hematopoietic stem cells. Ann N Y Acad Sci. 2012;1266:40–6.
Sun J, Ramos A, Chapman B, Johnnidis JB, Le L, Ho Y-J, et al. Clonal dynamics of native haematopoiesis. Nature. 2014;514:322–7.
Busch K, Klapproth K, Barile M, Flossdorf M, Holland-Letz T, Schlenner SM, et al. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature. 2015;518:542–6.
Sawai CM, Babovic S, Upadhaya S, Knapp DJHF, Lavin Y, Lau CM, et al. Hematopoietic stem cells are the major source of multilineage hematopoiesis in adult animals. Immunity. 2016;45:597–609.
Takizawa H, Boettcher S, Manz MG. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood. 2012;119:2991–3002.
Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-γ in response to chronic infection. Nature. 2010;465:793–7.
Burberry A, Zeng MY, Ding L, Wicks I, Inohara N, Morrison SJ, et al. Infection mobilizes hematopoietic stem cells through cooperative NOD-like receptor and toll-like receptor signaling. Cell Host Microbe. 2014;15:779–91.
Essers MAG, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, et al. IFNα activates dormant haematopoietic stem cells in vivo. Nature. 2009;458:904–8.
Sato T, Onai N, Yoshihara H, Arai F, Suda T, Ohteki T. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon–dependent exhaustion. Nat Med. 2009;15:696–700.
Manz MG, Boettcher S. Emergency granulopoiesis. Nat Rev Immunol. 2014;14:302–14.
Schmid MA, Takizawa H, Baumjohann DR, Saito Y, Manz MG. Bone marrow dendritic cell progenitors sense pathogens via Toll-like receptors and subsequently migrate to inflamed lymph nodes. Blood. 2011;118:4829–40.
Massberg S, Schaerli P, Knezevic-Maramica I, Köllnberger M, Tubo N, Moseman EA, et al. Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell. 2007;131:994–1008.
Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S, et al. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity. 2006;24:801–12.
Liu A, Wang Y, Ding Y, Baez I, Payne KJ, Borghesi L. Cutting edge: hematopoietic stem cell expansion and common lymphoid progenitor depletion require hematopoietic-derived, cell-autonomous TLR4 in a model of chronic endotoxin. J Immunol (Baltimore, Md.: 1950). 2015;195:2524–8.
Megías J, Yáñez A, Moriano S, O’Connor J-E, Gozalbo D, Gil M-L. Direct toll-like receptor-mediated stimulation of hematopoietic stem and progenitor cells occurs in vivo and promotes differentiation toward macrophages. Stem Cells. 2012;30:1486–95.
Zhao JL, Ma C, O'Connell RM, Mehta A, DiLoreto R, Heath JR, Baltimore D. Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress-induced hematopoiesis. Cell Stem Cell. 2014;14(4):445–59. doi:10.1016/j.stem.2014.01.007
Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature. 2010;465:793–7.
Walter D, Lier A, Geiselhart A, Thalheimer FB, Huntscha S, Sobotta MC, et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature. 2015;520:549–52.
Pietras EM, Passegué E. Linking HSCs to their youth. Nat Cell Biol. 2013;15:885–7.
Kitagawa M, Saito I, Kuwata T, Yoshida S, Yamaguchi S, Takahashi M, et al. Overexpression of tumor necrosis factor (TNF)-alpha and interferon (IFN)-gamma by bone marrow cells from patients with myelodysplastic syndromes. Leukemia. 1997;11:2049–54.
Ren G, Zhang L, Zhao X, Xu G, Zhang Y, Roberts AI, et al. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell. 2008;2:141–50.
Xie Z, Hao H, Tong C, Cheng Y, Liu J, Pang Y, et al. Human umbilical cord-derived mesenchymal stem cells elicit macrophages into an anti-inflammatory phenotype to alleviate insulin resistance in type 2 diabetic rats. Stem Cells. 2016;34:627–39.
Day RB, Bhattacharya D, Nagasawa T, Link DC. Granulocyte colony-stimulating factor reprograms bone marrow stromal cells to actively suppress B lymphopoiesis in mice. Blood. 2015;125:3114–7.
Schajnovitz A, Itkin T, D’Uva G, Kalinkovich A, Golan K, Ludin A, et al. CXCL12 secretion by bone marrow stromal cells is dependent on cell contact and mediated by connexin-43 and connexin-45 gap junctions. Nat Immunol. 2011;12:391–8.
Bunnell BA, Betancourt AM, Sullivan DE. New concepts on the immune modulation mediated by mesenchymal stem cells. Stem Cell Res Ther. 2010;1:34.
Martino MM, Maruyama K, Kuhn GA, Satoh T, Takeuchi O, Müller R, et al. Inhibition of IL-1R1/MyD88 signalling promotes mesenchymal stem cell-driven tissue regeneration. Nat Commun. 2016;7:11051.
Zhang WQ, Huang SH, Huang X, Li JH, Ye P, Xu J, Zheng PZ, Shen HY, Huang JR. Regulation of human mesenchymal stem cell differentiation by TREM-2. Hum Immunol. 2016:77(6):476–82. doi:10.1016/j.humimm.2015.06.005
Shi C, Jia T, Méndez-Ferrer S, Hohl TM, Serbina NV, Lipuma L, et al. Bone marrow mesenchymal stem and progenitor cells induce monocyte emigration in response to circulating toll-like receptor ligands. Immunity. 2011;34:590–601.
Wang J, Sun Q, Morita Y, Jiang H, Groß A, Lechel A, et al. A differentiation checkpoint limits hematopoietic stem cell self-renewal in response to DNA damage. Cell. 2012;148:1001–14.
Boettcher S, Ziegler P, Schmid MA, Takizawa H, van Rooijen N, Kopf M, et al. Cutting edge: lPS-induced emergency myelopoiesis depends on TLR4-expressing nonhematopoietic cells. J Immunol. 2012;188:5824–8.
Boettcher S, Gerosa RC, Radpour R, Bauer J, Ampenberger F, Heikenwalder M, et al. Endothelial cells translate pathogen signals into G-CSF-driven emergency granulopoiesis. Blood. 2014;124:1393–403.
Andonegui G, Zhou H, Bullard D, Kelly MM, Mullaly SC, McDonald B, Long EM, Robbins SM, Kubes P. Mice that exclusively express TLR4 on endothelial cells can efficiently clear a lethal systemic Gram-negative bacterial infection. J Clin Invest. 2009;119(7):1921–30
Broudy VC, Kaushansky K, Segal GM, Harlan JM, Adamson JW. Tumor necrosis factor type alpha stimulates human endothelial cells to produce granulocyte/macrophage colony-stimulating factor. Proc Natl Acad Sci USA. 1986;83:7467–71.
Sieff CA, Tsai S, Faller DV. Interleukin 1 induces cultured human endothelial cell production of granulocyte-macrophage colony-stimulating factor. J Clin Investig. 1987;79:48–51.
Fernandez L, Rodriguez S, Huang H, Chora A, Fernandes J, Mumaw C, et al. Tumor necrosis factor-α and endothelial cells modulate Notch signaling in the bone marrow microenvironment during inflammation. Exp Hematol. 2008;36:545–58.e1.
Luu NT, McGettrick HM, Buckley CD, Newsome PN, Rainger GE, Frampton J, et al. Crosstalk between mesenchymal stem cells and endothelial cells leads to downregulation of cytokine-induced leukocyte recruitment. Stem Cells (Dayton, Ohio). 2013;31:2690–702.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat Immunol. 2011;12:715–23.
Kristinsson SY, Landgren O, Samuelsson J, Bjorkholm M, Goldin LR. Autoimmunity and the risk of myeloproliferative neoplasms. Haematologica. 2010;95:1216–20.
Kristinsson SY, Bjorkholm M, Hultcrantz M, Derolf AR, Landgren O, Goldin LR. Chronic immune stimulation might act as a trigger for the development of acute myeloid leukemia or myelodysplastic syndromes. J Clin Oncol. 2011;29:2897–903.
Raaijmakers MHGP. Niche contributions to oncogenesis: emerging concepts and implications for the hematopoietic system. Haematologica. 2011;96:1041–8.
Raaijmakers MHGP, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464:852–7.
Zambetti NA, Ping Z, Chen S, Kenswil KJG, Mylona MA, Sanders MA, et al. Mesenchymal inflammation drives genotoxic stress in hematopoietic stem cells and predicts disease evolution in human pre-leukemia. Cell Stem Cell. 2016;19:613–27.
Takizawa H, Boettcher S, Manz MG. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood. 2012;119:2991–3002.
Haas S, Hansson J, Klimmeck D, Loeffler D, Velten L, Uckelmann H, et al. Inflammation-induced emergency megakaryopoiesis driven by hematopoietic stem cell-like megakaryocyte progenitors. Cell Stem Cell. 2015;17:422–34.
Kovtonyuk LV, Fritsch K, Feng X, Manz MG, Takizawa H. Inflamm-aging of hematopoiesis, hematopoietic stem cells, and the bone marrow microenvironment. Front Immunol. 2016;7:502.
Acknowledgements
This work was supported by KAKENHI from Japanese Society of the Promotion of Science (15H01519), KANAE Foundation for the Promotion of Medical Science, and SENSHIN Medical Research Foundation to H.T., and the Swiss National Science Foundation (310030_146528/1), the Promedica Foundation (Chur, Switzerland), and the Clinical Research Priority Program of the University of Zurich to M.G.M.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
About this article

Cite this article
Takizawa, H., Manz, M.G. Impact of inflammation on early hematopoiesis and the microenvironment. Int J Hematol 106, 27–33 (2017). https://doi.org/10.1007/s12185-017-2266-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-017-2266-5