
Abstract
Cellular and gene therapies represent promising treatment strategies at the frontier of medicine. Hematopoietic stem cells, lymphocytes, and mesenchymal stem cells (MSCs) can all serve as sources of cells for use in such therapies. Strategies for gene therapy are often based on those of cell therapy, and it is anticipated that some examples will be put to practical use in the near future. Given their ability to support hematopoiesis, MSCs may be useful for the enhancement of stem cell engraftment, and the acceleration of hematopoietic reconstitution. Furthermore, MSCs may advance the treatment of severe graft-versus-host disease, based on their immunosuppressive ability. This application is also based on the homing behavior of MSCs to sites of injury and inflammation. Interestingly, MSCs possess tumor-homing ability, opening up the possibility of applications in the targeted delivery of anti-cancer genes to tumors. Many reports have indicated that MSCs can be utilized to target tumors and to deliver anti-cancer molecules locally, as tumors are recognized as non-healing wounds with inflammatory tissue. Here, we review both the potential of MSCs as cellular vehicles for targeted cancer therapy and the molecular mechanisms underlying MSC accumulation at tumor sites.
Similar content being viewed by others


Introduction
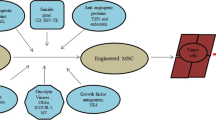
Mesenchymal stem cells (MSCs) are non-hematopoietic progenitor cells that can be easily isolated from a variety of tissues including bone marrow, adipose tissue, and the placenta/umbilical cord. They can be identified, expanded, genetically modified, and have the ability to differentiate into a variety of cell lineages, including adipocytes, osteocytes, chondrocytes, muscle cells, and stromal cells in vitro [1]. Recent studies have demonstrated that MSCs are capable of supporting hematopoiesis and can modulate immune responses. Interestingly, MSCs exhibit a homing behavior toward damaged tissue, inflammatory sites, and tumor sites (Fig. 1). Another feature of MSCs is their low immunogenicity because of a lack of expression of co-stimulatory molecules [2]. Therefore, MSCs do not activate the host immune response and escape immunological rejection when injected into HLA-non-identical recipients. These characteristics permit infusion of MSCs into recipients without HLA matching. The tumor site-homing activity of MSCs facilitates their use as cellular vehicles for the delivery of anti-cancer agents specifically to tumors [3]. This targeted therapy can reduce the systemic side effects of anti-cancer agents by facilitating their effective concentration at local tumor sites without elevating systemic concentrations. However, how and why MSCs accumulate at tumors remains poorly understood. Therefore, this review focuses on the application of MSCs for the targeted delivery of anti-cancer agents to tumors, and on the molecular mechanisms of their accumulation in these tumors.

Concept of regenerative medicine and cell therapy with MSCs. In bone marrow, there are different types of tissue stem cells including hematopoietic stem cells and MSCs. MSCs account for a small population of cells in bone marrow, and have the capacity to differentiate into a variety of mesenchymal lineages. Interestingly, MSCs have the ability to accumulate at the site of (1) damaged tissue; (2) inflammatory tissue; and (3) tumors. Therefore, MSCs can be utilized for: (1) regenerative therapy; (2) treatment of GVHD; (3) cancer gene therapy (targeted delivery)
Applications of genetically engineered MSCs for cancer therapy
An early application of MSCs to targeted cancer therapy was directed delivery of interferon beta (IFN-β). MSCs were transduced with the IFN-β gene, followed by their infusion into mice carrying melanoma xenografts. MSC treatment resulted in reduced tumor growth and prolonged survival of tumor-bearing mice [3]. Based on this observation, genetically modified MSCs from various tissues have since been evaluated for their therapeutic efficacy and their ability to act as cellular vehicles [3–23].
Interleukins (ILs) that regulate inflammatory and immune responses are often used as therapeutic agents. IL-12-expressing MSCs (MSC-IL-12) have been used to improve tumor immunological surveillance by activating cytotoxic lymphocytes and natural killer (NK) cells. Administration of MSC-IL-12 was shown to suppress metastasis and increase tumor cell apoptosis in mice bearing melanoma, lung cancer and hepatoma. Other immunomodulatory cytokines have also been examined for anti-tumor activities, including the T cell attractant CX3CL1. For example, MSC-CX3CL1 inhibited lung metastasis of melanoma and prolonged survival of tumor-bearing mice.
Suicide cancer gene therapy has been considered in the treatment of invasive tumors such as malignant glioma. A number of prodrug activation schemes that convert non-toxic prodrugs into toxic anti-metabolites are available for the selective killing of tumor cells. Cytosine deaminase (CD) and herpes simplex virus thymidine kinase (HSV-TK), which confer sensitivity to 5-Fluorocytosine (5-FC) and ganciclovir (GCV), respectively, are being evaluated in clinical trials. HSV-TK-expressing MSCs were injected in the vicinity of tumors, reducing tumor volume through bystander-mediated tumor cell killing following administration of GCV. Our group has developed genetically modified MSCs that produce retroviral vectors encoding HSV-TK, with the aim of augmenting the therapeutic efficacy of suicide cancer gene therapy (Fig. 2). MSCs isolated from the bone marrow of Sprague–Dawley rats were transfected with plasmid DNA expressing HSV-TK alone (non-VP-MSCs) or whole retroviral vector components (LTR-HSV-TK with Gag-pol and VSV-G; VP-MSCs) by nucleofection. To evaluate therapeutic efficacy, tumor-bearing nude mice were treated with non-VP-MSCs or VP-MSCs combined with GCV, and the size of subcutaneous tumors was periodically measured. Here, tumor growth was more efficiently suppressed by injection of VP-MSCs compared with non-VP-MSCs. This efficacy was dependent on therapeutic gene amplification through production of retrovirus progeny from MSCs and transduction of tumor cells in situ.

Left panel non-vector-producing MSCs (non-VP-MSCs). Although non-VP-MSCs have the ability to home to tumor sites, local expression of therapeutic molecules is dependent on the continued presence of the MSCs. Right panel vector-producing MSCs (VP-MSCs). The retrovirus progeny produced by MSCs can transduce tumor cells in situ, extending the expression of therapeutic molecules from tumor cells, even when the MSCs have died off
The efficacy of other therapeutic genes including IFN-α, cytosine deaminase (CD), NK4, and TRAIL has also been examined in various tumor models as described in Table 1. Furthermore, oncolytic virotherapy is emerging as a promising strategy for tumor treatment. MSCs can be employed as carriers to deliver oncolytic viruses to tumor sites. Experimentally, injection of genetically modified MSCs has resulted in tumor growth inhibition, metastasis suppression, and prolonged survival.
Interactions between tumors and MSCs
As detailed above, the use of MSCs to deliver anti-cancer agents is an attractive novel cancer therapeutic strategy. Endothelial cells (ECs), pericytes, and stromal cells are all known to support tumor growth and contribute to the tumor microenvironment by producing various growth factors including vascular endothelial growth factor (VEGF)-A, IL-8, transforming growth factor (TGF)-β, epidermal growth factor (EGF), and platelet-derived growth factor (PDGF) [24–29]. MSCs can also act in such a supportive fashion. It is speculated that when MSCs accumulate at tumor sites, they differentiate into pericytes or cancer-associated fibroblasts (CAFs) and are involved in facilitating tumor growth by providing structural support to the tumor microenvironment [3, 30, 31]. Interestingly, breast cancer cells stimulate MSCs and MSCs in turn support cancer cell invasion and metastasis by secreting CCL5 [32]. Conversely, MSCs can also induce apoptosis in tumor cells by blocking phosphorylation of AKT or preventing cell cycle progression [33]. Moreover, MSCs can produce DKK-1 to attenuate the potential for growth and malignancy of tumor cells [34]. As MSCs have dual and opposing effects with respect to tumor growth, modification with anti-tumor genes is required for MSC-based cancer-targeted therapy. Furthermore, it is also necessary to promote more efficient MSC accumulation at tumor sites.
Molecular mechanisms of MSC accumulation at tumors
If it becomes possible to increase the accumulation efficiency of MSCs at tumor sites, MSCs can effectively target not only primary tumors but also metastatic lesions. It is thought that MSCs are mobilized to damaged tissues, such as in injury or inflammation, by the release of inflammatory cytokines. Tumors possess a microenvironment consisting of a large number of inflammatory cells [35]. This microenvironment promotes the recruitment of MSCs via various soluble factors secreted by both tumor and inflammatory cells, including EGF, VEGF-A, PDGF, IL-8, IL-6, fibroblast growth factor, stromal cell-derived factor, granulocyte colony-stimulating factor, granulocyte–macrophage colony-stimulating factor, monocyte chemoattractant protein-1 (MCP-1), hepatocyte growth factor, TGF-β1, and urokinase-type plasminogen activator [36–42]. However, our own work found that while systemically injected MSCs accumulated at tumor sites, subcutaneously injected MSCs did not. We also compared the migration capacity of MSCs and fibroblasts (FBs) toward growth factors and chemokines in vitro, and found that FBs were more strongly attracted to these factors than MSCs [43]. These results suggest that the mechanism of MSC accumulation cannot be explained solely by cytokine-mediated migration.
The tumors generated in the above study possessed tumor stroma with many blood vessels, and MSCs in particular accumulated at the boundaries between tumors and tumor stroma. Furthermore, MSC accumulation at the tumor sites was observed only when cells were injected via the left ventricular cavity. Therefore, we focused on MSC-EC adhesion to elucidate the mechanisms involved. Interestingly, stimulation of MSCs with TNF-α enhanced the adhesion of MSCs to ECs in vitro. This adhesion was partially inhibited by antibodies that block vascular cell adhesion molecule-1 (VCAM-1) and very late antigen-4 (VLA-4). It is well known that TNF-α induces VCAM-1 expression via the NF-κB signaling pathway. Parthenolide (PTL) has anti-inflammatory activity and suppresses NF-κB activity by inhibiting IκBα phosphorylation after TNF-α stimulation, and PTL strongly inhibited TNF-α-induced VCAM-1 expression on MSCs. In vivo imaging using luciferase-expressing MSCs revealed that the bioluminescent signal gradually increased at tumor sites in mice injected with untreated MSCs. In contrast, we observed very weak signals at tumor sites in mice injected with PTL-treated MSCs. These results suggest that NF-κB activity regulates MSC accumulation at tumors by inducing VCAM-1 expression and subsequent cellular interaction with tumor vessel ECs (Fig. 3).

Proposed mechanism of MSC accumulation at tumors. Growth factors and chemokines recruit MSCs to the tumor microenvironment. Inflammatory cytokines, including TNF-α, stimulate MSCs and VCAM-1 expression is induced. Activated MSCs attach to the tumor vasculature, penetrate, and accumulate at tumor sites
Considerations for the use of genetically modified MSCs in cancer therapy
Although we focused on the function of TNF-α in the above study, other inflammatory cytokines including IL-1β and IFN-γ have the ability to induce VCAM-1 expression on target cells, and may also be involved in MSC accumulation. TNF-α is a major inflammatory cytokine that has important roles in diverse cellular events including cell survival, proliferation, differentiation, and death. Numerous reports have demonstrated elevated TNF-α levels in the serum of cancer patients, and TNF-α correlates closely with tumor progression and metastasis [44, 45]. For example, TNF-α readily induces IL-6 and MCP-1 secretion by CAFs and normal FBs and has an indirect influence on the generation of a prometastatic microenvironment. Furthermore, TNF-α is released in cardiac infarction [46] and graft-versus-host disease [47, 48]; MSCs accumulate at the site of cardiac infarction [49, 50]. These results indicate that pro-inflammatory cytokines also promote homing of MSCs in the heart and that these cytokines have a positive effect on cardiac regeneration. Therefore, MSC-based tissue-targeting strategies could be adapted for various inflammatory diseases, and activation with TNF-α may be one of the critically important steps for MSC accumulation.
For MSC-based cancer-targeted gene therapies, it is thought that therapeutic efficacy is directly coupled with the efficiency of MSC accumulation at tumor sites. Results from our laboratory suggest that the combined use of NF-κB inhibitors, including bortezomib, or TNF-α blocking agents, such as infliximab, reduces the therapeutic efficacy of genetically modified MSCs because of inhibition of MSC accumulation at the tumor. In contrast, tumor-specific TNF-α-inducing agents would be useful in enhancing therapeutic efficacy, thus further investigation is required for identifying such agents to establish more effective therapeutic strategies.
Conclusion
The application of anti-cancer gene-expressing MSCs for targeted cancer therapy is a novel and promising strategy. Here, we propose that suicide cancer gene therapy may be improved using vector-producing MSCs. This strategy is likely to generate vectors in situ, leading to the killing of solid tumors. This could be achieved using MSCs to initiate virus production near tumor cells in situ. These viruses are then transduced into tumor cells, which themselves produce virus progeny, thereby amplifying the transgene expression at tumor sites. While the therapeutic benefit and safety of this approach requires further examination, it holds great potential for the eradication of tumors.
MSC accumulation at tumor sites is related to migratory capacity toward growth factors and chemokines and also MSC-EC adhesion following activation by TNF-α. Furthermore, NF-κB activity regulates MSC accumulation at tumor sites through the induction of VCAM-1 expression and the resultant interaction with tumor blood vessel ECs. Although MSCs are useful as cellular vehicles for cancer-targeted gene therapy, previous studies have shown that increased MSC accumulation is required to enhance therapeutic efficacy. Thus, mechanisms of enhancing MSC accumulation should be developed, and the TNF-α-NF-κB-VCAM-1 axis may represent a solution to this problem.
References
Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–7.
Nauta AJ, Fibbe WE. Immunomodulatory properties of mesenchymal stromal cells. Blood. 2007;110:3499–506.
Studeny M, Marini FC, Champlin RE, Zompetta C, Fidler IJ, Andreeff M. Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer Res. 2002;62:3603–8.
Ren C, Kumar S, Chanda D, Chen J, Mountz JD, Ponnazhagan S. Therapeutic potential of mesenchymal stem cells producing interferon-alpha in a mouse melanoma lung metastasis model. Stem Cells. 2008;26:2332–8.
Studeny M, Marini FC, Dembinski JL, Zompetta C, Cabreira-Hansen M, Bekele BN, et al. Mesecnhymal stem cells: potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. J Natl Cancer Inst. 2004;96:1593–603.
Ren C, Kumar S, Chanda D, Kallman L, Chen J, Mountz JD, et al. Cancer gene therapy using mesenchymal stem cells expressing interferon-β in a mouse prostate cancer lung metastasis model. Gene Ther. 2008;15:1446–53.
Nakamizo A, Marini F, Amano T, Khan A, Studeny M, Gumin J, et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005;65:3307–18.
Li X, Lu Y, Huang W, Xu H, Chen X, Geng Q, et al. In vitro effect of adenovirus-mediated human Gamma Interferon gene transfer into human mesenchymal stem cells for chronic myelogenous leukemia. Hematol Oncol. 2006;24:151–8.
Nakamura K, Ito Y, Kawano Y, Kurozumi K, Kobune M, Tsuda H, et al. Antitumor effect of genetically engineered mesenchymal stem cells in a rat glioma model. Gene Ther. 2004;11:1155–64.
Chen X, Lin X, Zhao J, Shi W, Zhang H, Wang Y, et al. A tumor-selective biotherapy with prolonged impact on established metastases based on cytokine gene-engineered MSCs. Mol Ther. 2008;16:749–56.
Chen XC, Wang R, Zhao X, Wei YQ, Hu M, Wang YS, et al. Prophylaxis against carcinogenesis in three kinds of unestablished tumor models via IL12-gene-engineered MSCs. Carcinogenesis. 2006;27:2434–41.
Xin H, Kanehira M, Mizuguchi H, Hayakawa T, Kikuchi T, Nukiwa T, et al. Targeted delivery of CX3CL1 to multiple lung tumors by mesenchymal stem cells. Stem Cells. 2007;25:1618–26.
Uchibori R, Okada T, Ito T, Urabe M, Mizukami H, Kume A, et al. Retroviral vector-producing mesenchymal stem cells for targeted suicide cancer gene therapy. J Gene Med. 2009;11:373–81.
Matuskova M, Hlubinova K, Pastorakova A, Hunakova L, Altanerova V, Altaner C, et al. HSV-tk expressing mesenchymal stem cells exert bystander effect on human glioblastoma cells. Cancer Lett. 2009;290:58–67.
Kucerova L, Matuskova M, Pastorakova A, Tyciakova S, Jakubikova J, Bohovic R, et al. Cytosine deaminase expressing human mesenchymal stem cells mediated tumour regression in melanoma bearing mice. J Gene Med. 2008;10:1071–82.
Kucerova L, Altanerova V, Matuskova M, Tyciakova S, Altaner C. Adipose tissue-derived human mesenchymal stem cells mediated prodrug cancer gene therapy. Cancer Res. 2007;67:6304–13.
Kanehira M, Xin H, Hoshino K, Maemondo M, Mizuguchi H, Hayakawa T, et al. Targeted delivery of NK4 to multiple lung tumors by bone marrow-derived mesenchymal stem cells. Cancer Gene Ther. 2007;14:894–903.
Komarova S, Kawakami Y, Stoff-Khalili MA, Curiel DT, Pereboeva L. Mesenchymal progenitor cells as cellular vehicles for delivery of oncolytic adenoviruses. Mol Cancer Ther. 2006;5:755–66.
Hakkarainen T, Särkioja M, Lehenkari P, Miettinen S, Ylikomi T, Suuronen R, et al. Human mesenchymal stem cells lack tumor tropism but enhance the antitumor activity of oncolytic adenoviruses in orthotopic lung and breast tumors. Hum Gene Ther. 2007;18:627–41.
Stoff-Khalili MA, Rivera AA, Mathis JM, Banerjee NS, Moon AS, Hess A, et al. Mesenchymal stem cells as a vehicle for targeted delivery of CRAds to lung metastases of breast carcinoma. Breast Cancer Res Treat. 2007;105:157–67.
Loebinger MR, Eddaoudi A, Davies D, Janes SM. Mesenchymal stem cell delivery of TRAIL can eliminate metastatic cancer. Cancer Res. 2009;69:4134–42.
Kim SM, Lim JY, Park SI, Jeong CH, Oh JH, Jeong M, et al. Gene therapy using TRAIL-secreting human umbilical cord blood-derived mesenchymal stem cells against intracranial glioma. Cancer Res. 2008;68:9614–23.
Sasportas LS, Kasmieh R, Wakimoto H, Hingtgen S, van de Water JA, Mohapatra G, et al. Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proc Natl Acad Sci USA. 2009;106:4822–7.
Benjamin LE, Golijanin D, Itin A, Pode D, Keshet E. Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J Clin Invest. 1999;103:159–65.
Song S, Ewald AJ, Stallcup W, Werb Z, Bergers G. PDGFR beta+ perivascular progenitor cells in tumours regulate pericyte differentiation and vascular survival. Nat Cell Biol. 2005;7:870–9.
Bababeygy SR, Cheshier SH, Hou LC, Higgins DM, Weissman IL, Tse VC. Hematopoietic stem cell-derived pericytic cells in brain tumor angioarchitecture. Stem Cells Dev. 2008;17:11–8.
Lyden D, Hattori K, Dias S, Costa C, Blaikie P, Butros L, et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7:1194–201.
Peters BA, Diaz LA, Polyak K, Meszler L, Romans K, Guinan EC, et al. Contribution of bone marrow-derived endothelial cells to human tumor vasculature. Nat Med. 2005;11:261–2.
Spring H, Schüler T, Arnold B, Hämmerling GJ, Ganss R. Chemokines direct endothelial progenitors into tumor neovessels. Proc Natl Acad Sci USA. 2005;102:18111–6.
Bexell D, Gunnarsson S, Tormin A, Darabi A, Gisselsson D, Roybon L, et al. Bone marrow multipotent mesenchymal stroma cells act as pericyte-like migratory vehicles in experimental gliomas. Mol Ther. 2009;17:183–90.
Bexell D, Scheding S, Bengzon J. Toward brain tumor gene therapy using multipotent mesenchymal stromal cell vectors. Mol Ther. 2010;18:1067–75.
Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–63.
Etheridge SL, Spencer GJ, Heath DJ, Genever PG. Expression profiling and functional analysis of wnt signaling mechanisms in mesenchymal stem cells. Stem Cells. 2004;22:849–60.
Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7.
Tille JC, Pepper MS. Mesenchymal cells potentiate vascular endothelial growth factor-induced angiogenesis in vitro. Exp Cell Res. 2002;280:179–91.
Andrades JA, Han B, Becerra J, Sorgente N, Hall FL, Nimni ME. A recombinant human TGF-beta1 fusion protein with collagen-binding domain promotes migration, growth, and differentiation of bone marrow mesenchymal cells. Exp Cell Res. 1999;250:485–98.
Yu J, Ustach C, Kim HR. Platelet-derived growth factor signaling and human cancer. J Biochem Mol Biol. 2003;36:49–59.
Wang L, Li Y, Chen X, Chen J, Gautam SC, Xu Y, et al. MCP-1, MIP-1, IL-8 and ischemic cerebral tissue enhance human bone marrow stromal cell migration in interface culture. Hematology. 2002;7:113–7.
Wang L, Li Y, Chen J, Gautam SC, Zhang Z, Lu M, et al. Ischemic cerebral tissue and MCP-1 enhance rat bone marrow stromal cell migration in interface culture. Exp Hematol. 2002;30:831–6.
Rempel SA, Dudas S, Ge S, Gutiérrez JA. Identification and localization of the cytokine SDF1 and its receptor, CXC chemokine receptor 4, to regions of necrosis and angiogenesis in human glioblastoma. Clin Cancer Res. 2000;6:102–11.
Honczarenko M, Le Y, Swierkowski M, Ghiran I, Glodek AM, Silberstein LE. Human bone marrow stromal cells express a distinct set of biologically functional chemokine receptors. Stem Cells. 2006;24:1030–41.
Ponte AL, Marais E, Gallay N, Langonné A, Delorme B, Hérault O, et al. The in vitro migration capacity of human bone marrow mesenchymal stem cells: comparison of chemokine and growth factor chemotactic activities. Stem Cells. 2007;25:1737–45.
Uchibori R, Tsukahara T, Mizuguchi H, Saga Y, Urabe M, Mizukami H, et al. NF-κB activity regulates mesenchymal stem cell accumulation at tumor sites. Cancer Res. 2013;73:364–72.
Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer. 2009;9:361–71.
Wu Y, Zhou BP. TNF-alpha/NF-kappaB/snail pathway in cancer cell migration and invasion. Br J Cancer. 2010;102:639–44.
Shpektor A. Cardiogenic shock: the role of inflammation. Acute Card Care. 2010;12:115–8.
Levine JE. Implications of TNF-α in the pathogenesis and management of GVHD. Acute Card Care. 2010;12:115–8.
Hao L, Sun H, Wang J, Wang T, Wang M, Zou Z. Mesenchymal stromal cells for cell therapy: besides supporting hematopoiesis. Int J Hematol. 2012;95:34–46.
Shake JG, Gruber PJ, Baumgartner WA, Senechal G, Meyers J, Redmond JM, et al. Mesenchymal stem cell implantation in a swine myocardial infarct model: engraftment and functional effects. Ann Thorac Surg. 2002;73:1919–25.
Pittenger MF, Martin BJ. Mesenchymal stem cells and their potential as cardiac therapeutics. Circ Res. 2004;95:9–20.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Uchibori, R., Tsukahara, T., Ohmine, K. et al. Cancer gene therapy using mesenchymal stem cells. Int J Hematol 99, 377–382 (2014). https://doi.org/10.1007/s12185-014-1537-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-014-1537-7