
Abstract
Adoptive transfer of tumor-reactive T cells into cancer patients with the intent of inducing a cytotoxic anti-tumor effector response and durable immunity has long been proposed as a novel therapy for a broad range of malignancies; however, local and systemic tolerance mechanisms have hindered the generation of effective T cell therapies and limited the clinical efficacy of this approach in cancer patients. Chimeric antigen receptors (CARs) are recombinant receptors that comprise an extracellular antigen-targeting domain in conjunction with one or more intracellular T cell signaling domains that can be introduced into T cells by genetic modification to redirect their specificity to the CAR-targeted antigen. Administration of CD19-specific CAR-modified T cells that target B cell non-Hodgkin lymphomas and leukemia has been remarkably effective in recent clinical trials, energizing the field and stimulating new efforts to identify the critical parameters of CAR design and T cell engineering that are necessary for effective cancer therapy.
Similar content being viewed by others

Introduction
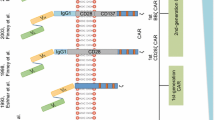
The observation that adaptive immunity plays a critical role in surveillance and control of cancer spurred great hopes that the exquisite specificity of T cell effector functions, directed by the interaction between the T cell receptor (TCR) and its cognate MHC-peptide complex, could be harnessed to target malignant tumors [1]. Adoptive immunotherapy strategies were developed to allow collection of T cells from the blood or tumor tissue of a patient with cancer, manipulation of harvested T cells in vitro to generate an expanded population of tumor-reactive T cells, and infusion of tumor-reactive T cells back into the patient with the intent of inducing a cytotoxic anti-tumor effector response and establishing durable immunity to prevent subsequent relapse of the malignancy (Fig. 1) [2–7]. Careful comparative analyses of non-responding patients and those with impressive (albeit sporadic) clinical responses have allowed the field to continue to mature since the first studies of T cell therapy for cancer were conducted. The observation that clinical responses were uncommon in patients in whom tumor-reactive T cells could not be detected in vivo after in vitro expansion and adoptive transfer demonstrated the importance of T cell persistence after adoptive transfer [7] and stimulated investigation into strategies to define the qualities of transferred T cells and the recipient microenvironment that are necessary for T cell persistence and clinical efficacy. Although sporadic impressive remissions were seen in studies of antigen-stimulated T cell or tumor-infiltrating lymphocyte (TIL) infusions, durable clinical responses were uncommon, in part because high-avidity T cells that target self-antigens expressed by tumors were likely eliminated during thymic negative selection and could not be generated for T cell therapy. The recent development of efficient methods to genetically modify T cells provided a means by which T cells that were not subject to negative selection could be redirected to target self-antigens on tumor tissue, thus overcoming the obstacles imposed by central tolerance. The recent notable success in the treatment of patients with B cell malignancies using genetically redirected T cells has energized the field and ignited extraordinary enthusiasm for the future of adoptive T cell therapy [8, 9].

Generation of tumor-specific T cells by repeated antigen stimulation or genetic modification to express a tumor-targeting receptor. PBMC collected from a patient or healthy individual can be stimulated in vitro with tumor antigen at regular intervals to induce gradual enrichment of antigen-specific T cells (blue). Multiple stimulations followed by additional enrichment or expansion strategies are required to ensure sufficient antigen-specific T cells are generated. The entire process may take 2–3 months. In contrast, approaches that utilize genetic modification to redirect T cell specificity to a tumor antigen are much more rapid. PBMC can be collected from a patient or healthy donor and retrovirally or lentivirally transduced to express a tumor-reactive CAR (or TCR). The enriched CAR-modified tumor-reactive T cells (red) can be infused into the patient in as little as 1–2 weeks
Redirecting T cell specificity by genetic modification
The majority of reported antigens that have been targeted by T cell adoptive therapy are self-antigens expressed by normal tissues in addition to the tumor. Negative selection ensures that T cells that have high avidity for self-antigens are deleted in the thymus, averting the potential for autoimmunity due to self-reactive T cells. However, the corollary is that self-reactive T cells that escape thymic deletion are likely of low avidity and may not be optimal for targeting T cells to tumors. To circumvent the lack of high-avidity tumor reactive T cells, high-affinity tumor-specific receptors can be introduced by genetic modification into T cells that comprise the native repertoire and were not subject to central tolerance. Redirection of T cell specificity to a tumor can be accomplished by introduction of transgenes encoding α and β chains that encode a TCR specific for a tumor antigen or by introduction of tumor antigen-specific chimeric antigen receptors (CARs) [10, 11]. These strategies endow modified T cells with the capacity to be activated through the introduced antigen receptor or their endogenously expressed TCR; hence their designation in some studies as bi-specific T cells. CARs generally comprise a tumor-targeting domain, a spacer, a transmembrane domain, and one or more intracellular T cell signaling domains. The most commonly used approaches to introduce CARs into T cells involve gamma retroviral or lentiviral transduction; however, other strategies such as the Sleeping Beauty system and electroporation of naked DNA plasmid or mRNA have been employed [12–16]. CAR-modified T cells can be activated by any surface-expressed antigen to which a scFv can be generated, rather than being restricted to TCR-mediated recognition of short peptide antigens that are processed and presented by HLA molecules. scFv-based redirection of specificity enables a high-avidity interaction with the target cell, with a lower risk of off-target or degenerate recognition that may be problematic after TCR-modification due to mispairing of endogenous and introduced TCR α and β chains [17]. Because CAR-mediated T cell activation is not dependent on HLA molecule expression, CARs can be used to target T cells to tumors in patients with different HLA haplotypes. CAR-modified T cells are also impervious to some of the immune escape mechanisms that impair the interaction of a conventional TCR with a tumor-derived peptide-HLA complex, such as modification of tumor antigen processing and downregulation of HLA molecule expression. The incorporation of costimulatory molecule signaling sequences in the CAR construct adds another barrier to immune escape due to downregulation of costimulatory ligand expression on tumor cells.
Design of chimeric antigen receptors
While CAR design has evolved from the early constructs described in 1993 when T cell activation was achieved by stimulation through a chimeric receptor comprising a murine single chain variable fragment (scFv) and CD3ζ or FcRγ [18], the utilization of a scFv derived from a monoclonal antibody to a cell surface antigen has remained the most frequently employed CAR targeting strategy [19]. Multiple cell surface antigens expressed by B cell malignancies, including CD19, CD20, CD22, CD23, CD38, ROR1, and kappa light chain, have been targeted with T cells engineered to express scFv-containing CAR constructs [13, 15, 20–25], and some of these antigens are now being targeted in phase I clinical trials of CAR-modified T cell therapy (Table 1). Although targeting using a scFv incorporated into a CAR has remained the most prevalent strategy to target B cell malignancies, other approaches have been used to target CAR-modified T cells to solid tumors, for example by incorporation of IL-13 into a CAR to redirect engineered T cells to IL-13Rα2-expressing tumors [26].
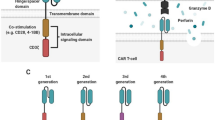
Receptor design has proceeded in an iterative fashion from the first generation of CARs that incorporated a scFv and T cell stimulatory domain (Fig. 2) [18]. Subsequent studies demonstrated that activation of T cells engineered to express first-generation CARs was incomplete and that T cells modified with second- or third-generation CARs that incorporated, respectively, one or more intracellular costimulatory signaling domains derived from CD28, 41BB, OX40, DAP10, ICOS, or CD27 possessed greater proliferative capacity, cytokine secretion, and anti-tumor activity than their first generation counterparts [27–34]. The importance of costimulation was confirmed in a clinical trial in which patients with B cell non-Hodgkin lymphoma were treated with T cells modified to express a CD19-specific CAR that incorporated either a CD28 costimulatory domain or no costimulation [35]. Greater in vivo T cell proliferation and persistence was seen after infusion of T cells modified with the second-generation CAR incorporating CD3ζ and a CD28 costimulatory domain compared with those modifed with the first-generation CD3ζ CAR without costimulation. While there is general agreement that T cells modified to express second-generation CARs are more effective than those engineered to express their first-generation counterparts, there is no consensus about the selection of costimulatory molecule domain to incorporate into second-generation CARs or whether third-generation CARs are superior to second-generation CARs [36, 37]. One clinical trial has investigated infusion of T cells modified to express a third-generation CD20-specific CAR that incorporated costimulatory domains derived from both CD28 and 41BB [15]; however, clinical efficacy and T cell persistence were modest. It is possible that the strategies used for engineering and prolonged culture of the CAR-modified T cells in this study may have limited their in vivo persistence and efficacy; thus additional work will be needed before definitive conclusions about the use of third-generation CARs can be drawn.

Chimeric antigen receptor design. a Three generations of CAR design are depicted. First-generation CARs comprise an extracellular antigen receptor, most often a scFv, and an intracellular T cell signaling domain, usually CD3ζ. Second- and third-generation CARs incorporate in addition one or two costimulatory domains, respectively. b A first-generation CAR can be engineered into the same cell in conjunction with another CAR that incorporates a unique scFv and a costimulatory domain, but no CD3ζ signaling domain. This allows robust activation of the T cell to occur only after encounter with two distinct antigens. EC extracellular, Cyto cytoplasm, TM transmembrane
While T cells modified to express a CAR respond to ligation of either the CAR or their naïve TCR, the impact of signaling through a CAR instead of a TCR has not been completely characterized [13]. The greater affinity of a scFv for target antigen compared with the affinity of a native TCR for its cognate peptide-MHC complex may be advantageous in promoting high-avidity interaction between the modified T cell and tumor, as supported by the finding that targeting of ROR1+ cell lines was more effective with T cells engineered to express a CAR incorporating a high-affinity ROR1-specific scFv compared with a low-affinity scFv [38]. However, the effects of the high-affinity CAR-target interaction on formation of the immune synapse, propagation of signaling, and gene transcription have not been determined. Other aspects of CAR design, such as the characteristics of the spacer and transmembrane domains or the location of the target epitope may also affect the outcome of T cell activation. For example, a ROR1-specific CAR incorporating a short extracellular spacer between the scFv and transmembrane domain was more effective than its long spacer counterpart in lysis of ROR1+ tumor cells [38], and CAR-mediated targeting of an epitope of CD22 located in close proximity to the tumor cell membrane was more efficient than targeting a more distally located epitope [39]. The density of target molecules on the tumor cell and of CARs on the T cell may also significantly impact CAR-modified T cell function [39, 40]. These observations highlight the fact that CAR design is in evolution, and it is presently unclear if there are cardinal characteristics of CAR design that are necessary for optimal clinical efficacy of CAR-modified T cells. It is evident that the requirements for CAR design may differ between CARs targeting distinct antigens, and other factors, such as the T cell subset that expresses the CAR, may also dictate aspects of optimal CAR structure [41]. Evaluation of CAR signaling, structural modeling of CAR-ligand interactions, combinatorial analyses of CAR designs, and analyses of in vivo efficacy in animal models will provide insight and guide future approaches to CAR-modified T cell engineering.
In addition to improvements in CAR design, other approaches have been investigated to improve CAR-modified T cell function and persistence. While costimulation has most often been incorporated within the CAR construct, T cell function can be augmented by provision of costimulation in formats that do not include the costimulatory domain within the CAR [42]. Engineering T cells to express both the CAR and costimulatory molecule ligands such as CD80 or CD86 may ensure delivery of a costimulatory signal to adoptively transferred T cells in the absence of tumor-derived costimulation [42]. Another approach involves modifying T cells to express both a first-generation CAR and a separate chimeric receptor that contains a distinct extracellular antigen-binding domain and a costimulatory domain, but no CD3ζ signaling domain (Fig. 2). This strategy may restrict full activation of the modified T cells to those that encounter target cells that express two distinct tumor antigens. Addition of transgenes encoding IL-2, IL-7, IL-15 or IL-21 has been studied in an effort to improve the effector function, persistence or in vivo efficacy of CAR-modified T cells [43]; however, the finding of persistent autonomous proliferation of a CD8+ T cell clone transduced to express IL-15 suggests that caution is warranted in translation of this approach [44]. A similar approach, which involves introduction of a transgene encoding IL-12 with the CAR construct, has been investigated as a strategy to overcome the suppressive effects of regulatory CD4+ Foxp3+ T cells (Tregs) in the tumor microenvironment [45]. Other investigators have attempted to circumvent the problem of suppression by Tregs by modifying CAR-engineered T cells to express a transgene encoding IL7Rα, which confers the CAR-engineered T cells with a selective growth advantage when cultured in IL-7 compared with Tregs that do not express IL-7Rα [46].
Composition of CAR-modified T cell products
The human CD4+ and CD8+ T cell pools contain distinct subsets that differ in frequency, phenotype, transcriptional and epigenetic profiles, and function [47–51], and these different attributes of distinct T cell subsets may affect their suitability for CAR engineering and adoptive immunotherapy. Central memory CD8+ T cells from non-human primates that are stimulated and cultured in vitro then re-infused can be detected in blood, lymph nodes, and bone marrow for beyond 1 year after adoptive transfer, whereas their counterparts generated after stimulation and culture of effector memory CD8+ T cells cannot be detected [52]. These data suggest that central memory CD8+ T cells may be better for adoptive immunotherapy than effector memory cells. Other studies have suggested that naïve T cells or recently described memory stem cells might be optimal for adoptive T cell therapy [50, 53].
A reductionist approach that involves isolation of distinct effective T cell subsets prior to CAR engineering is a logical option, but it remains unknown if it is necessary. CAR engineering of unselected PBMC or T cells will modify distinct subsets contained within a leukapheresis product or blood sample, including those that might be required for synergistic activity, and this approach has produced exciting data in clinical trials [8, 9]. However, a resounding question is whether CAR modification and infusion of unselected PBMC or T cells result in transfer of subsets that might be associated with undue toxicity or those, such as Tregs, that could inhibit an anti-tumor response. Because the composition of T cell subsets in blood is highly variable between individuals of different ages and with distinct histories of antigen encounter and exposure to chemotherapy, and distinct subsets may proliferate in culture at different rates, separate culture, and formulation of CAR-modified T generated from distinct T cell subsets may provide a way forward that excludes deleterious subsets and allows synergy between effective subsets. Studies of CD4+ and CD8+ central memory CAR T cells that are separately stimulated, transduced, and cultured prior to formulation at a defined ratio for infusion are currently in progress.
Clinical trials
CAR-modified T cells are emerging as an exciting new therapeutic reagent for patients with B cell malignancies (Table 1) and potentially for patients with other cancers. While many potential antigens that enable targeting of B cell malignancies have been identified, the B lineage-restricted expression of CD19 and its presence on most B cell leukemias and lymphomas have made it the preferred target for a majority of phase I studies of CAR-modified T cell therapy [8, 9, 12, 35, 54–60], many of which have demonstrated remarkable efficacy in patients with leukemias and lymphomas that were resistant to conventional therapy.
At the University of Pennsylvania, 2 of 3 patients with chronic lymphocytic leukemia (CLL) who received lymphodepleting chemotherapy and autologous T cells modified to express a second generation CD19-specific CAR that incorporated a 4-1BB costimulatory domain and CD3ζ signaling domain achieved durable complete remissions [9, 55]. After adoptive transfer, in vivo proliferation of CAR-modified T cells and delayed tumor lysis syndrome were reported. At the same center, two patients with B cell acute lymphoblastic leukemia (B-ALL) also achieved complete remission after CD19-specific CAR-modified autologous T cell therapy; however, one relapsed within 2 months of therapy with CD19-negative disease [59]. Other studies in which patients were treated with autologous T cells modified with CD19-specific CARs that incorporated costimulatory domains derived from CD28 have also demonstrated anti-tumor effects in patients with B cell malignancies [8, 54, 56, 57]. Remarkable efficacy was seen after treatment of 5 B-ALL patients at Memorial Sloan Kettering Cancer Center with lymphodepleting chemotherapy and T cells modified to express a CD19-specific CAR incorporating a CD28 costimulatory domain [8]. Two recent reports have suggested that CD19-specific CAR-modified T cell therapy may have anti-tumor activity in allogeneic hematopoietic stem cell transplant (HCT) recipients [54, 60]. The concern that administration of allogeneic CAR-modified T cells that express endogenous potentially alloreactive TCRs could exacerbate graft versus host disease (GVHD) stimulated development of strategies to CAR-modify virus-specific T cells, which have been shown to have a lower risk of causing GVHD after adoptive transfer [13, 54, 61]. However, in the limited number of patients treated to date, therapy with allogeneic CAR-modified unselected T cells was not associated with severe GVHD [54, 60].
Autologous CAR-modified T cells engineered to target CD20 have also been tested in phase I clinical trials [15, 20, 58]; however, in these early studies CD20-specific CAR-modified T cells showed modest efficacy compared with that reported in more recent clinical trials of CD19-targeted CAR-modified T cell therapy. The development of new strategies for CAR design and improved methods of T cell transduction and culture suggest that re-investigation of targeting of CD20 should be considered.
Anti-tumor activity in humans has been noted after therapy with T cells that were engineered to express CARs that differ in scFv, spacer domain, transmembrane domain, and costimulatory molecule selection, and with CAR-modified T cells that were isolated, transduced, and cultured under different conditions. At this early stage, few definite conclusions can be drawn regarding the optimal approach to CAR-modified T cell generation, and future studies to compare CAR design and critical cell processing variables are anticipated.
Toxicity of CAR-modified T cells
Most of the serious reported toxicities of CD19-specific CAR-modified T cells have been ‘on-target’ toxicities that occur as a result of the recognition of the target antigen, CD19, by the CAR-modified T cells. On encounter with antigen in vivo after adoptive transfer, CD19-specific CAR-modified T cells proliferate and accumulate in the recipient, leading to rapid elimination of tumor [9]. Tumor lysis syndrome with severe renal impairment has developed in patients treated with CD19-specific CAR-modified T cells, in some cases at remarkably late times after T cell infusion [9, 62]. Cytokine release syndrome, manifested by fever and hypotension occurring days to weeks after T cell infusion, has been noted as a consequence of cytokine secretion in response to antigen-mediated activation of CD19-specific CAR-modified T cells [59]. Macrophage activation syndrome is a distinct clinical entity that occurs after infusion of CD19-specific CAR-modified T cell infusion and is associated with delayed fever, hemophagocytosis, hyperferritinemia, and elevation of serum IL-6 [59]. One reported patient who was critically ill with macrophage activation syndrome after infusion of CD19-specific CAR-modified T cells demonstrated a dramatic response to therapy with tocilizumab, an anti-IL-6R monoclonal antibody [59]. The pathogenic mechanisms that lead to macrophage activation syndrome after CAR-modified T cell therapy have not been defined.
Both normal and malignant B cells express CD19; therefore, an expected on-target (but off-tumor) toxicity of CD19-specific CAR-modified T cell therapy is depletion of endogenous B lymphocytes [9, 57]. Although B lymphocyte depletion has been profound in reported clinical studies, its durability has not been completely defined and is likely to in part depend on the persistence of the infused T cells. While B lymphocyte depletion increases the risk of opportunistic infection, this may be ameliorated by replacement intravenous immunoglobulin therapy. In addition, strategies to eliminate transferred T cells from patients with complete tumor responses will likely be investigated in future studies to enable endogenous B lymphocyte recovery.
Strategies to improve the safety of CAR-modified T cells
While CD19-specific CAR-modified T cell therapy has shown impressive anti-tumor activity in early clinical studies, the potential for serious adverse events due to on-target toxicity is considerable, suggesting that future incorporation of strategies to eliminate transferred T cells is warranted. Suicide genes may be incorporated into CAR-encoding vectors. Thymidine kinase from HSV (HSV-TK) has been used as an effective suicide gene in transferred T cells, but its immunogenicity may limit its future utility in adoptive T cell transfer [58, 63]. Recent studies demonstrated that administration of a small molecule dimerizer (AP1903) to allogeneic HCT recipients induced rapid amelioration of acute GVHD and elimination of transplanted T cells that were engineered to express an AP1903-inducible caspase [64]. An alternative strategy that is in development involves engineering of CAR-modified T cells with a truncated human epidermal growth factor (EGF) receptor (EGFR) that lacks the EGF-binding domain and the intracellular signaling domain, but retains the extracellular epitope to which the clinically available anti-EGFR monoclonal antibody, cetuximab (Erbitux), binds, potentially allowing the use of systemic administration of cetuximab as a strategy to deplete engineered EGFR+ T cells [65].
Future directions
As T cell therapy advances beyond early phase clinical trials, the next challenges will be to formulate strategies that improve cell isolation and culture and allow large-scale automation of CAR-modified T cell production, reduce the expense of engineered T cell therapies, and facilitate their delivery beyond the academic research environment. Despite the early success of therapy of B cell malignancies with T cells modified to target CD19, it remains unknown whether similar results will be achieved by targeting other antigens expressed by B cell malignancies or antigens expressed by other tumors; careful research at the bench and bedside to define the characteristics of effective CAR-modified T cell preparations and facilitate the design and clinical application of CAR-modified T cell therapies will ensure these questions are addressed.
References
Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 2007;450:903–7.
Turtle CJ, Hudecek M, Jensen MC, Riddell SR. Engineered T cells for anti-cancer therapy. Curr Opin Immunol. 2012;24:633–9.
Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci USA. 2002;99:16168–73.
Chapuis AG, Thompson JA, Margolin KA, Rodmyre R, Lai IP, Dowdy K, et al. Transferred melanoma-specific CD8+ T cells persist, mediate tumor regression, and acquire central memory phenotype. Proc Natl Acad Sci USA. 2012;109:4592–7.
Chapuis AG, Ragnarsson GB, Nguyen HN, Chaney CN, Pufnock JS, Schmitt TM, et al. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Sci Transl Med. 2013;5:174ra27.
Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med. 2008;358:2698–703.
Robbins PF, Dudley ME, Wunderlich J, El-Gamil M, Li YF, Zhou J, et al. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol. 2004;173:7125–30.
Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra38.
Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced Leukemia. Sci Transl Med. 2011;3:95ra73.
Schmitt TM, Ragnarsson GB, Greenberg PD. T cell receptor gene therapy for cancer. Hum Gene Ther. 2009;20:1240–8.
Turtle CJ, Riddell SR. Genetically retargeting CD8+ lymphocyte subsets for cancer immunotherapy. Curr Opin Immunol. 2011;23:299–305.
Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–28.
Terakura S, Yamamoto TN, Gardner RA, Turtle CJ, Jensen MC, Riddell SR. Generation of CD19-chimeric antigen receptor modified CD8+ T cells derived from virus-specific central memory T cells. Blood. 2012;119:72–82.
Hackett PB, Largaespada DA, Cooper LJ. A transposon and transposase system for human application. Mol Ther. 2010;18:674–83.
Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood. 2012;119:3940–50.
Yoon SH, Lee JM, Cho HI, Kim EK, Kim HS, Park MY, et al. Adoptive immunotherapy using human peripheral blood lymphocytes transferred with RNA encoding Her-2/neu-specific chimeric immune receptor in ovarian cancer xenograft model. Cancer Gene Ther. 2009;16:489–97.
Kuball J, Dossett ML, Wolfl M, Ho WY, Voss RH, Fowler C, et al. Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood. 2007;109:2331–8.
Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA. 1993;90:720–4.
Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3:388–98.
Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA, et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 2008;112:2261–71.
Hudecek M, Schmitt TM, Baskar S, Lupo-Stanghellini MT, Nishida T, Yamamoto TN, et al. The B-cell tumor-associated antigen ROR1 can be targeted with T cells modified to express a ROR1-specific chimeric antigen receptor. Blood. 2010;116:4532–41.
Giordano Attianese GM, Marin V, Hoyos V, Savoldo B, Pizzitola I, Tettamanti S, et al. In vitro and in vivo model of a novel immunotherapy approach for chronic lymphocytic leukemia by anti-CD23 chimeric antigen receptor. Blood. 2011;117:4736–4745.
Mihara K, Yanagihara K, Takigahira M, Kitanaka A, Imai C, Bhattacharyya J, et al. Synergistic and persistent effect of T-cell immunotherapy with anti-CD19 or anti-CD38 chimeric receptor in conjunction with rituximab on B-cell non-Hodgkin lymphoma. Br J Haematol. 2010;151:37–46.
Vera J, Savoldo B, Vigouroux S, Biagi E, Pule M, Rossig C, et al. T lymphocytes redirected against the kappa light chain of human immunoglobulin efficiently kill mature B lymphocyte-derived malignant cells. Blood. 2006;108:3890–7.
Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121:1165–74.
Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004;64:9160–6.
Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta/CD28 receptor. Nat Biotechnol. 2002;20:70–5.
Song DG, Ye Q, Carpenito C, Poussin M, Wang LP, Ji C, et al. In vivo persistence, tumor localization, and antitumor activity of CAR-engineered T cells is enhanced by costimulatory signaling through CD137 (4-1BB). Cancer Res. 2011;71:4617–27.
Song DG, Ye Q, Poussin M, Harms GM, Figini M, Powell DJJ. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood. 2012;119:696–706.
Finney HM, Akbar AN, Lawson AD. Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J Immunol. 2004;172:104–13.
Wilkie S, Picco G, Foster J, Davies DM, Julien S, Cooper L, et al. Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J Immunol. 2008;180:4901–9.
Hombach AA, Abken H. Costimulation by chimeric antigen receptors revisited the T cell antitumor response benefits from combined CD28-OX40 signalling. Int J Cancer. 2011;129:2935–44.
Pule MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther. 2005;12:933–41.
Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N, et al. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006;66:10995–1004.
Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822–6.
Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci USA. 2009;106:3360–5.
Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther. 2010;18:413–20.
Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin Cancer Res. 2013;19:3153–64.
James SE, Greenberg PD, Jensen MC, Lin Y, Wang J, Till BG, et al. Antigen sensitivity of CD22-specific chimeric TCR is modulated by target epitope distance from the cell membrane. J Immunol. 2008;180:7028–38.
James SE, Greenberg PD, Jensen MC, Lin Y, Wang J, Budde LE, et al. Mathematical modeling of chimeric TCR triggering predicts the magnitude of target lysis and its impairment by TCR downmodulation. J Immunol. 2010;184:4284–94.
Hombach AA, Chmielewski M, Rappl G, Abken H. Adoptive immunotherapy with redirected T cells produces CCR7- cells that are trapped in the periphery and benefit from combined CD28-OX40 costimulation. Hum Gene Ther. 2013;24:259–69.
Stephan MT, Ponomarev V, Brentjens RJ, Chang AH, Dobrenkov KV, Heller G, et al. T cell-encoded CD80 and 4-1BBL induce auto- and transcostimulation, resulting in potent tumor rejection. Nat Med. 2007;13:1440–9.
Markley JC, Sadelain M. IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell-mediated rejection of systemic lymphoma in immunodeficient mice. Blood. 2010;115:3508–19.
Hsu C, Jones SA, Cohen CJ, Zheng Z, Kerstann K, Zhou J, et al. Cytokine-independent growth and clonal expansion of a primary human CD8+ T-cell clone following retroviral transduction with the IL-15 gene. Blood. 2007;109:5168–77.
Lee JC, Hayman E, Pegram HJ, Santos E, Heller G, Sadelain M, et al. In vivo inhibition of human CD19-targeted effector T cells by natural T regulatory cells in a xenotransplant murine model of B cell malignancy. Cancer Res. 2011;71:2871–81.
Perna SK, Pagliara D, Mahendravada A, Liu H, Brenner MK, Savoldo B, et al. Interleukin-7 mediates selective expansion of tumor-redirected cytotoxic T lymphocytes without enhancement of regulatory T-cell inhibition. Clin Cancer Res. 2013.
Araki Y, Wang Z, Zang C, Wood Iii WH, Schones D, Cui K, et al. Genome-wide analysis of histone methylation reveals chromatin state-based regulation of gene transcription and function of memory CD8+ T cells. Immunity. 2009;30:912–25.
Weng NP, Araki Y, Subedi K. The molecular basis of the memory T cell response: differential gene expression and its epigenetic regulation. Nat Rev Immunol. 2012;12:306–15.
Turtle CJ, Swanson HM, Fujii N, Estey EH, Riddell SR. A distinct subset of self-renewing human memory CD8 + T cells survives cytotoxic chemotherapy. Immunity. 2009;31:834–44.
Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17:1290–7.
Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–12.
Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118:294–305.
Hinrichs CS, Borman ZA, Gattinoni L, Yu Z, Burns WR, Huang J, et al. Human effector CD8+ T cells derived from naive rather than memory subsets possess superior traits for adoptive immunotherapy. Blood. 2011;117:808–14.
Cruz CR, Micklethwaite KP, Savoldo B, Ramos CA, Lam S, Ku S, et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood. 2013;122:2965–73.
Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–33.
Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–102.
Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–20.
Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16:1245–56.
Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–18.
Kochenderfer JN, Dudley ME, Carpenter RO, Kassim SH, Rose JJ, Telford WG, et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood. 2013 [Epub ahead of print].
Melenhorst JJ, Leen AM, Bollard CM, Quigley MF, Price DA, Rooney CM, et al. Allogeneic virus-specific T cells with HLA alloreactivity do not produce GVHD in human subjects. Blood. 2010;116:4700–2.
Kalos M, June CH. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity. 2013;39:49–60.
Berger C, Flowers ME, Warren EH, Riddell SR. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood. 2006;107:2294–302.
Di Stasi SK, Tey A, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–83.
Wang X, Chang WC, Wong CW, Colcher D, Sherman M, Ostberg JR, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118:1255–63.
Acknowledgments
The author acknowledges support from the National Institutes of Health (1K99CA154608-01A1, 4R00CA154608-03) and is a 2013 Damon Runyon Clinical Investigator.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Turtle, C.J. Chimeric antigen receptor modified T cell therapy for B cell malignancies. Int J Hematol 99, 132–140 (2014). https://doi.org/10.1007/s12185-013-1490-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-013-1490-x