
Abstract
The discovery of activating mutations in JAK2 and MPL in a majority of patients with myeloproliferative neoplasms (MPN) has led to the rapid clinical development of several JAK kinase inhibitors. Of these, the JAK1/2 inhibitor, ruxolitinib (INCB018424, Incyte Corporation) was recently approved for the treatment of patients with myelofibrosis (MF). JAK inhibitors have effectively reduced splenomegaly and high cytokine levels in patients leading to improvements in quality of life. However, they have not been successful in eliminating the mutant clone in a majority of patients. In vitro studies using saturation mutagenesis screens have revealed several mutations in JAK2 that confer resistance to JAK inhibitors. Nevertheless, these mutations have not been identified so far in JAK inhibitor-treated patients. A recent study from our laboratory demonstrated that chronic JAK kinase inhibition leads to JAK inhibitor persistence via transphosphorylation of JAK2 through other JAK kinase family members. This phenomenon is seen in cell lines, mouse models and patient samples. The JAK inhibitor persistent cells, however, still remain JAK2 dependent and therefore combination therapies that target JAK2 and other components of the JAK–STAT pathway along with JAK inhibitors may provide additional benefits and improve clinical outcomes in these patients.
Similar content being viewed by others

The spectrum of myeloproliferative neoplasms
The World Health Organization (WHO) classifies patients with myeloproliferative neoplasms (MPN) into two categories, those that are Philadelphia (Ph+) chromosome positive with classical chronic myeloid leukemia (CML), and those patients that are Philadelphia chromosome negative (Ph−). This review will focus on the non-CML classical MPN that includes the clinically distinct diseases polycythemia vera, essential thrombocytosis (ET) and primary myelofibrosis (PMF). It was the foresight of the American hematologist, William Dameshek, who, in 1951 [1], suggested that these diseases were closely interrelated manifestations of abnormal bone marrow proliferation.
In patients, PV manifests itself with an excessive proliferation of three cell lineages: erythroid, myeloid and megakaryocytic cells. Patients typically present with increased numbers of erythrocytes accompanied by increase in hematocrit and hemoglobin levels, with variable leukocytosis and thrombocytosis. They often have splenomegaly and their bone marrows may have increased reticulin fibers indicative of progressive fibrosis. Secondary myelofibrosis can occur in roughly 6 % of PV patients with an increased risk of progression over time [2]. ET patients have an extremely high platelet count with a normal red blood cell count, and as in PV and at risk for thrombotic and hemorrhagic events. Post-ET myelofibrosis is less common than post-PV myelofibrosis, but does occur in a subset of ET patients. PMF patients, on the other hand, present with extensive bone marrow fibrosis, anemia, extramedullary hematopoiesis, organomegaly and an altered cytokine expression profile.
Mutations in the JAK–STAT pathway
It took more than 50 years after Dameshek’s seminal observations for investigators to uncover the first molecular culprit for these disorders. In 2005, several groups reported the discovery of a somatic activating mutation in JAK2 in a majority of MPN patients, thereby vindicating Dameshek’s observations [3–6]. JAK2 is a member of the Janus family of non-receptor tyrosine kinases that mediate downstream JAK–STAT signaling upon activation by a cognate cytokine. The JAK2V617F mutation occurs in 90–99 % of PV patients, 41–72 % of ET patients and 39–57 % of PMF patients, making it the most frequently found mutation thus far in MPN patients. The majority of the small subset of PV patients who did not harbor JAK2V617F were found to bear mutations in another region of JAK2, specifically in the SH2 domain inside exon 12 [7]. Further research led to the identification of MPLW515L/K mutations in a small percentage of ET and PMF patients (8 %), although the mutation frequency of MPL is less well characterized as for JAK2V617F [8]. However, regardless of the type of mutation, the hallmark of MPN pathogenesis is aberrant JAK–STAT signaling. Of note, mutations in several epigenetic regulators have been observed recently in MPN patients (mostly at a frequency < 10 %) including CBL, IDH1, IDH2, TET2, EZH2, DNMT3A, ASXL1, SF3B1, IKZF1 and others [9]; however, the role of these mutant disease alleles in MPN pathogenesis is beyond the scope of this review.
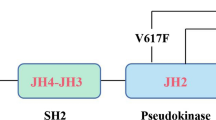
The Janus family of kinases is involved in the transduction of cytokine-mediated signals in a number of cell types and regulates cytokine-dependent gene expression, in part by activating the signal transducers and activators of transcription signaling effectors (STATs). Four mammalian JAKs have been identified, namely JAK1, JAK2, JAK3, and TYK2 [10–13]. These proteins are characterized by the presence of the kinase domain (JH1) along with a non-catalytic pseudokinase domain (JH2), which is postulated to serve as a negative regulator of JAK kinase activity [14]. Interestingly, the JAK2V617F mutation is located in the pseudokinase domain of JAK2 and recent studies have demonstrated that the pseudokinase domain has dual-specificity kinase activity, with the ability to phosphorylate two negative regulatory sites in JAK2: Ser523 and Tyr570 [15]. MPN patients that are JAK2V617F positive have reduced phosphorylation of JAK2 at Tyr570, suggesting that loss of JH2 activity contributes to the pathogenesis of MPN.
JAK1, JAK2 and TYK2 are ubiquitously expressed as compared to JAK3, which is primarily expressed in hematopoietic cells [16]. Upon ligand binding, cytokine receptor subunits associate, thereby resulting in homotypic or heterotypic dimerization of the JAK molecules that are associated with them. This results in transphosphorylation and initiation of signaling via recruitment and phosphorylation of the STAT family of transcription factors [13]. Upon getting activated by JAK kinases, the STATs can dimerize via their SH2 domains, which leads to nuclear translocation, binding to specific DNA sequences and transcriptional activation/repression of target genes [17]. Of note, the JAK–STAT pathway can interact with the receptor tyrosine kinase/Ras/MAPK pathway and also result in activation of the PI3K signaling pathway leading to complex biological consequences.
Recent data, however, has also shown that JAK2 can directly enter the nucleus and phosphorylate histone H3 at Tyr41 residue [18]. Resultant activation of H3 prevents its binding with HP1a, which in turn, causes repression of heterochromatin genes. Understanding this non-canonical activation of JAK2 will be important in delineating the emerging role of JAK2 in epigenetic regulation in MPN pathogenesis and other contexts.
Clinical experience with JAK inhibitors
Following the discovery of JAK mutations in a majority of MPN patients, JAK inhibitors entered clinical trials fairly rapidly. There are several JAK inhibitors in various stages of clinical development including, but not limited to INCB018424, SAR302503, CYT387, SB1518, CEP701, LY2784544, NS018, AZD1480, and BMS911543 (Table 1). The initial goal was to obtain rapid clinical and molecular responses, as have been observed using imatinib and other Abl kinase inhibitors for Ph+ CML patients. These JAK inhibitors differ in their structure and their specificity to JAK2 kinase. The most advanced amongst these inhibitors is the FDA approved agent ruxolitinib, also known as INCB018424.
Ruxolitinib
Ruxolitinib is an orally bioavailable, JAK1/2 kinase inhibitor that has been approved for treating high-risk PMF patients. It inhibits JAK1 and JAK2 at IC50 values of 3.3 and 2.8 nM, respectively, and has much lower affinity for TYK2 and JAK3 in cell-free assay systems [19]. Ruxolitinib potently inhibits JAK–STAT signaling in Ba/F3 cells engineered to express JAK2V617F, induces apoptosis in these cells and reduces erythroid colony formation in ex vivo-treated PV samples. Preclinical studies using ruxolitinib were highly encouraging with reduction in spleen size and normalization of cytokine profiles in treated mice [20].
Several excellent reviews have covered the clinical efficacy of ruxolitinib [20, 21]. Briefly, the Phase II/III COMFORT (COntrolled MyeloFibrosis study with ORal JAK inhibiTor) trials, in which patients were given ruxolitionib compared to placebo (COMFORT-I) or best available care (COMFORT-II) led to the FDA approval of ruxolitinib. These studies showed that ruxolitinib resulted in remarkable improvement in constitutional symptoms of patients, reduction in spleen sizes and abatement of the “cytokine storm” seen in these patients [22–26]. Ruxolitinib overall, seems to be well tolerated in patients with very few grade 3–4 side effects [24]. MF patients taking ruxolitinib had modest, albeit significant improvements in overall survival, possibly by improving cachexia, and some reversal in bone marrow fibrosis [25]. Further, preliminary data from a Phase II study of ruxolitinib in PV patients also suggests significant reductions in white blood counts, improvement in constitutional symptoms and modest reduction in allele burden [27].
SAR302503
SAR302503 from Sanofi (formerly from TargeGen, TG101348) is another JAK2 inhibitor that has shown promise in clinical trials. TG101348 inhibits JAK2 at an IC50 of 3 nM and JAK1 at 105 nM, suggesting it is more JAK2 specific than ruxolitinib [28]. Further, the compound inhibited ex vivo hematopoietic colony growth in MPN patients [28] and was efficacious in a mouse bone marrow transplant driven by JAK2V617F [29]. The clinical experience with SAR302503 in a Phase II study showed improvements in splenomegaly and constitutional symptoms, and durable, albeit modest, reduction in mutant allele burdens in a subset patients with intermediate or high-risk myelofibrosis, PV, or ET, and in patients with ruxolitinib-resistant or intolerant myelofibrosis [30]. SAR302503 is also being investigated in a Phase III trial in patients with intermediate-2 or high-risk myelofibrosis, and the results from that clinical trial should be available soon.
CYT387
A recent report at the American Society of Hematology annual meeting presented data on an extended Phase II trial of CYT387 (from Gilead Sciences, formerly called Cytopia) [31]. CYT387 is a Type I JAK1/2 inhibitor with IC50 values of 11 and 18 nM, respectively, for JAK1 and JAK2 [32]. Along with improvements in patient constitutional symptoms and reduction in splenomegaly, about 70 % of the patients enrolled in this trial became transfusion independent for prolonged periods suggesting this agent may have different effects on erythroid response compared to other agents in this class.
Lestaurtinib
Lestaurtinib or CEP701 (Cephalon) has been used in clinical trials for AML patients based on its anti-Flt3 activity [33]. The compound also inhibits JAK2 at an IC50 concentration of 0.9 nM [34]. Lestaurtinib could also inhibit JAK–STAT signaling in erythroid precursor cells from MPN patients. A Phase II clinical study of CEP701 in JAK2V617F-positive PMF or post-PV/ET myelofibrosis patients showed modest efficacy with no improvement in bone marrow fibrosis or mutant allele burden [35].
Pacritinib
Pacritinib or SB1518 (SBio) is a pyrimidine-based JAK inhibitor that has much higher efficacy against JAK2 (IC50 = 19–23 nM) compared to other JAK kinases [36]. The compound also shows significant Flt3 activity (IC50 = 22 nM) and has been well tolerated in a Phase II study for MF patients [37].
Several other JAK inhibitors including LY2784544 [38], NS018 [39], AZD1480 [40], and BMS911543 [41] have been evaluated in Phase I/II clinical trials but there is little data available regarding the efficacy of these inhibitors thus far (Table 1).
How effective are JAK inhibitors as monotherapeutic agents?
All the JAK inhibitors tested thus far in clinical trials have been ATP mimetic Type I JAK inhibitors, which are defined by their ability to bind JAK2 in and around the region occupied by the adenine ring of ATP and do not require the DFG motif in the activation loop to adopt a ‘DFG-out’ conformation for binding. Treatment with Type I JAK inhibitors leads to paradoxical phosphorylation of JAK2, possibly a result of stabilization of activated JAK2 [42]. Further, compared to the dramatic effects of imatinib in CML, histopathologic and molecular responses in terms of allele burden reduction have been modest with JAK inhibitors. Also, unlike imatinib, where a maximum tolerated dose (MTD) was not attained for CML, treating MPN patients with JAK inhibitors leads to considerable side effects including anemia and thrombocytopenia, possibly due to the indispensable role of JAK2 in erythropoiesis [43].
The clinical experience with JAK inhibitors is similar to what has been observed in mouse preclinical models. Mullally et al. [44] generated a JAK2V617F knock-in mouse that had disease features of human PV. Treatment of the primary JAK2V617F knock-in mice with TG101348 for 6 weeks reduced spleen weights and improved histopathology in inhibitor-treated mice compared to vehicle-treated mice. The authors then purified LSKs from the vehicle-treated and JAK inhibitor-treated primary mice and transplanted equal number of cells into lethally irradiated secondary recipients. All the secondary recipients, however, showed complete hematological reconstitution along with increased hematocrits, suggesting that inhibitor treatment was not effective in eradicating or even reducing the number of MPN-initiating cells. Further, longer treatment duration of 10 weeks was also not enough to eliminate the disease initiating cells as seen by increased hematocrits in tertiary recipients 3 weeks after transplantation suggesting that JAK inhibitor therapy was not curative in this model. We have seen similar results in our MPLW515L GFP-driven mouse bone marrow transplant models that mimic many features of human ET/PMF [45]. We saw that while 4 weeks of treatment with INCB016562, another JAK2 inhibitor, reduced blood counts, improved survival and histopathology of treated mice, it did not reduce GFP percentage (a measure of the mutant allele burden) in peripheral blood or the proportion of GFP-positive HSC-enriched Lin−c-kit+sca-1+ (LSK) cells in treated mice.
Mechanisms of resistance to JAK2 inhibitors
Since JAK2 inhibitors have shown limited efficacy in reducing allele burden and fibrosis in MPN, several groups conducted in vitro studies to identify possible genetic mechanisms underlying this phenomenon. A saturation mutagenesis screen performed in JAK2V617 mutant cells identified five non-synonymous mutations in the JAK2 kinase domain that conferred resistance to ruxolitinib [46]. Further, these mutations displayed cross-resistance to other JAK2 kinase inhibitors such as CYT387, TG101348, CEP701 and AZD1480. To date these mutations have not been observed in the clinical setting.
Another group isolated several other mutations in TEL-JAK2 mutant cells, in which JAK2 is constitutively activated via the fusion of its pseudokinase and kinase domain to the PNT oligomerization domain of TEL [47]. These alterations primarily conferred resistance to JAK Inhibitor I, a commercially available pan-JAK inhibitor but did not affect response to other clinical inhibitors indicating that these might be compound specific mutations. Neither group isolated the putative gatekeeper mutations at position M929, which is predicted to confer resistance to ATP-competitive inhibitors, suggesting that these screens did not achieve complete saturation. Upon testing the gatekeeper mutation, they found it conferred modest resistance to ruxolitinib and JAK Inhibitor I.
Since JAK2 is mutated in other hematological malignancies including B-ALL, Weigert et al. [48] utilized a similar approach in JAK2R683G mutant cell lines using a novel JAK2 inhibitor, NVP-BVB808 and identified the same alleles as in previous studies. They also demonstrated that these alterations conferred varying degrees of resistance to other clinically relevant JAK inhibitors in JAK2V617F mutant cells. Table 2 summarizes all the mutations identified in various screens that have been carried out using JAK inhibitors in MPN.
All the mutations identified so far are located in the kinase domain of JAK2. A number of the mutations occur at residues located in the ATP binding pocket of JAK2 that have been shown to interact with JAK Inhibitor I based on the crystallographic analyses [49] and presumably would interact with other Type I JAK inhibitors (Fig. 1). They are also relatively few in number compared to those identified in BCR-ABL mutant cells in response to imatinib treatment [50] suggesting that a few critical residues are probably involved in mediating resistance in the future. However, as discussed above, to date second-side mutations in JAK2 have not been identified in MPN patients treated with JAK2 inhibitors. This might be due to dose-limiting toxicities associated with JAK2 inhibitors due to its critical role in normal hematopoiesis that do not allow achievement of high enough doses to induce genetic resistance.

Mechanisms of resistance to JAK2 inhibitors: a in naïve cells, JAK2 inhibitors are able to bind JAK2 and inhibit downstream STAT signaling. b Secondary mutations in JAK2 prevent the binding of inhibitors to the ATP binding pocket resulting in activation of STATs and expression of genes involved in proliferation and survival. c Prolonged exposure to JAK inhibitors can stabilize activated JAK2 and facilitate recruitment of other JAK kinases, which can transphosphorylate JAK2, thereby restoring downstream signaling pathways
We recently demonstrated that chronic exposure of MPN cells to ruxolitinib leads to the development of persistent cells in which JAK2 is activated via the formation of heterodimers with other JAK kinases including JAK1 and TYK2 [51]. These cells are cross-resistant to other JAK inhibitors including JAK Inhibitor I and TG101348. This phenotype was neither due to the outgrowth of a pre-existing persistent subpopulation nor due to acquisition of secondary mutations. We saw this phenomenon in cell lines, murine models, as well as in primary samples from patients treated with ruxolitinib. The underlying mechanism is based on the stabilization of activated JAK2 by the binding of Type I inhibitors, which facilitates the recruitment of other JAK kinases, which can then transactivate JAK2 and reactivate downstream signaling. Persistence is reversible and cells rapidly become resensitized following withdrawal of drug. This suggests that MPN patients might benefit from taking a drug holiday before being retreated with the same or different JAK2 inhibitor.
The JAK inhibitor persistent cells, however, remain dependent on JAK2, as knockdown by short hairpins targeting JAK2 inhibits their growth and inhibits downstream signaling. They also remain sensitive to Hsp90 inhibitors such as the novel purine-based PU-H71 that result in degradation of total JAK2. These data indicate that JAK2 is required, presumably as a signaling scaffold, even when its kinase activity is inhibited, and that degradation of the JAK2 kinase rather than inhibition of its kinase activity may have improved efficacy in MPN patients. Of note, PU-H71 causes lineage-specific reduction in myeloproliferation and is selectively retained in mutant cells, where it is able to degrade JAK2 in a tumor-selective manner [52]. Another strategy to target these persistent cells was the use of Type II inhibitors such as BBT-594, which bind JAK2 in a conformation-independent manner and do not contribute to stabilization of activated JAK2 [42].
Kalota et al. [53] reported that granulocytes from myelofibrosis patients are relatively insensitive to ex vivo JAK2 inhibition in terms of reduction in levels of phosphorylated STAT3 and STAT5 as compared to patients with PV and ET as well as normal controls. This was a cell-intrinsic phenomenon and was observed in JAK2 inhibitor-naïve patients suggesting that certain subgroups of patients might have de novo mechanisms of resistance/persistence to JAK inhibitors, which should be further investigated.
Alternative approaches using combinatorial therapy
Preclinical and clinical studies conducted to date indicate that JAK2 enzymatic inhibitors will likely not be curative as monotherapy in MPN. Therefore, a number of groups are investigating the potential of targeting alternative pathways in combination with JAK–STAT in disease models. JAK2 is a client protein of the Hsp90 chaperone complex and is rapidly degraded upon inhibition of Hsp90 by small molecules such as PU-H71 and AUY922 [48, 52]. Combining these inhibitors with JAK2 inhibitors was highly effective in inhibiting growth and survival in MPN cell lines, murine models and primary cells [48, 52, 54]. Hsp90 inhibition was also able to overcome persistence as well as genetic resistance to JAK2 inhibitors in cell lines and xenograft models [48, 51, 54]. These studies provide a compelling rationale for pursuing this avenue of combination therapies in the clinic.
HDAC inhibitors like panobinostat and givinostat also result in degradation of JAK2 and inhibition of growth and downstream signaling, possibly by disrupting the binding of JAK2 and HSP90 and through additional mechanisms relating to the pleiotropic role of HDAC proteins in MPN cells. In separate Phase I/II trials with panobinostat or givinostat, MF patients experienced improvement in systematic symptoms and reduction in splenomegaly [55–57]. Cotreatment of MPN cells with the JAK2 inhibitor TG101209 and panobinostat led to synergistic induction of apoptosis in MPN cells [58]. Similarly, combination of panobinostat with ruxolitinib showed greater activity than either agent alone in a JAK2V617 murine model [59]. There is an ongoing Phase I trial assessing the efficacy of combined ruxolitinib and panobinostat treatment.
In addition to enhanced JAK–STAT signaling, MPN cells display activation of other oncogenic pathways including MAPK and mTOR/PI3K signaling. In a Phase I/II trial evaluating the efficacy of the mTOR inhibitor, everolimus, 60 % of MF patients had improvement in constitutional symptoms and decrease in spleen enlargement albeit to a lesser degree than observed with JAK inhibitors. However, it did not lead to a decrease in mutant allele burden or significant changes in the cytokine profile of these patients [60]. Treatment of cultured as well as primary MPN cells with the dual PI3K/mTOR inhibitor BEZ235 combined with the JAK inhibitor SAR302503 had synergistic effects on induction of apoptosis and inhibition of colony growth in cultured and primary MPN cells as compared to normal CD34+ cells [61]. BEZ235 was also effective against a cell line that had been made resistant to TG101209 [61]. Similar results were also reported with the combination of JAK2 inhibitors with a MEK inhibitor, AZD6244 [62, 63].
Recently, developmental pathways such as Hedgehog, Wnt and Notch have been shown to play a role in development of myeloid malignancies and remain an active area of research as possible therapeutic targets [64–67]. In Phase I trials of the Hh inhibitor PF-04449913 in hematological malignancies including MPN, 4/5 MF patients attained stable disease while 1 experienced a clinical response including a reduction in spleen size [68]. Further studies are warranted to evaluate the efficacy of combining these agents with JAK kinase inhibitors.
In conclusion, JAK inhibitors improve constitutional symptoms and cachexia seen in MF patients. However, they are unable to render cytogenetic or molecular remission. Using allosteric JAK inhibitors or combination with other classes of inhibitors including HDAC and/or Hsp90 inhibitors or with inhibitors that target thePI3K/Akt/MEK pathways may provide further benefit without additional toxicities and help improve outcomes in these patients. Although JAK inhibitors represent a major milestone in improving outcomes for MPN patients, we believe that combination studies and informed development of second-generation JAK targeting therapies have the potential to more substantively improve outcomes for MPN patients.
References
Dameshek W. Some speculations on the myeloproliferative syndromes. Blood. 1951;6(4):372–5.
Passamonti F, et al. A dynamic prognostic model to predict survival in post-polycythemia vera myelofibrosis. Blood. 2008;111(7):3383–7.
Baxter EJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–61.
Levine RL, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–97.
James C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–8.
Kralovics R, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–90.
Scott LM, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356(5):459–68.
Pikman Y, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3(7):e270.
Shih AH, et al. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 2012;12(9):599–612.
Wilks AF, et al. Two novel protein-tyrosine kinases, each with a second phosphotransferase-related catalytic domain, define a new class of protein kinase. Mol Cell Biol. 1991;11(4):2057–65.
Ziemiecki A, Harpur AG, Wilks AF. JAK protein tyrosine kinases: their role in cytokine signalling. Trends Cell Biol. 1994;4(6):207–12.
Harpur AG, et al. JAK2, a third member of the JAK family of protein tyrosine kinases. Oncogene. 1992;7(7):1347–53.
Ihle JN. The Janus kinase family and signaling through members of the cytokine receptor superfamily. Proc Soc Exp Biol Med. 1994;206(3):268–72.
Saharinen P, Silvennoinen O. The pseudokinase domain is required for suppression of basal activity of Jak2 and Jak3 tyrosine kinases and for cytokine-inducible activation of signal transduction. J Biol Chem. 2002;277(49):47954–63.
Ungureanu D, et al. The pseudokinase domain of JAK2 is a dual-specificity protein kinase that negatively regulates cytokine signaling. Nat Struct Mol Biol. 2011;18(9):971–6.
Kawamura M, et al. Molecular cloning of L-JAK, a Janus family protein-tyrosine kinase expressed in natural killer cells and activated leukocytes. Proc Natl Acad Sci. 1994;91(14):6374–8.
Shuai K, et al. Polypeptide signalling to the nucleus through tyrosine phosphorylation of Jak and Stat proteins. Nature. 1993;366(6455):580–3.
Dawson MA, et al. JAK2 phosphorylates histone H3Y41 and excludes HP1[agr] from chromatin. Nature. 2009;461(7265):819–22.
Quintás-Cardama A, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115(15):3109–17.
Apostolidou E, Kantarjian HM, Verstovsek S. JAK2 inhibitors: a reality? A hope? Clin Lymphoma Myeloma. 2009;9(Suppl 3):S340–5. doi:10.3816/CLM.2009.s.033.
Pardanani A, et al. JAK inhibitor therapy for myelofibrosis: critical assessment of value and limitations. Leukemia. 2011;25(2):218–25.
Mesa RA, et al. Effect of ruxolitinib therapy on myelofibrosis-related symptoms and other patient-reported outcomes in COMFORT-I: a randomized, double-blind. placebo-controlled trial. J Clin Oncol. 2013;31(10):1285–92.
Verstovsek S, et al. Long-term outcomes of 107 patients with myelofibrosis receiving JAK1/JAK2 inhibitor ruxolitinib: survival advantage in comparison to matched historical controls. Blood. 2012;120(6):1202–9.
Verstovsek S, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799–807.
Verstovsek S, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 Inhibitor. myelofibrosis. New Engl J Med. 2010;363(12):1117–27.
Cervantes F, Kiladjian J-J, Niederwieser D, Sirulnik A, Stalbovskaya V, McQuity M, Hunter DS, Levy RS, Passamonti F, Barbui T, Barosi G, Gisslinger H, Vannucchi AM, Knoops L, Harrison CN. Long-term safety, efficacy, and survival findings from comfort-II, a Phase 3 study comparing ruxolitinib with best available therapy (BAT) for the treatment of myelofibrosis (MF). Blood. 2012;120:801 (ASH Annual Meeting Abstracts 2012).
Verstovsek S, Passamonti F, Rambaldi A, Barosi G, Rosen PJ, He S, Contel N, Mookerjee B, Rumi E, Gattoni E, Pieri L, Cazzola M, Kantarjian HM, Barbui T, Vannucchi AM. Long-term efficacy and safety results from a Phase II study of ruxolitinib in patients with polycythemia vera. Blood. 2012;120:804 (ASH Annual Meeting Abstracts 2012).
Lasho TL, et al. TG101348, a JAK2-selective antagonist, inhibits primary hematopoietic cells derived from myeloproliferative disorder patients with JAK2V617F, MPLW515K or JAK2 exon 12 mutations as well as mutation negative patients. Leukemia. 2008 (published online).
Wernig G, et al. Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera. Cancer Cell. 2008;13(4):311–20.
Pardanani A, et al. Safety and Efficacy of TG101348, a Selective JAK2 Inhibitor. Myelofibrosis. J Clin Oncol. 2011;29(7):789–96.
Pardanani A, Gotlib J, Gupta V, Roberts AW, Wadleigh M, Sirhan S, Bavisotto LM, Kawashima J, Kowalski M, Tefferi A Phase I/II Study of CYT387, a JAK1/JAK2 Inhibitor for the treatment of myelofibrosis. Blood. 2012;120:178 (ASH Annual Meeting Abstracts 2012).
Pardanani A, et al. cyt387, a selective jak1/jak2 inhibitor: in vitro assessment of kinase selectivity and preclinical studies using cell lines and primary cells from polycythemia vera patients. Leukemia. 2009;23(8):1441–5.
Smith BD, et al. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004;103(10):3669–76.
Hexner EO, et al. Lestaurtinib (CEP701) is a JAK2 inhibitor that suppresses JAK2/STAT5 signaling and the proliferation of primary erythroid cells from patients with myeloproliferative disorders. Blood. 2008;111(12):5663–71.
Santos FPS, et al. Phase 2 study of CEP-701, an orally available JAK2 inhibitor, in patients with primary or post-polycythemia vera/essential thrombocythemia myelofibrosis. Blood. 2010;115(6):1131–6.
Hart S, et al. SB1518, a novel macrocyclic pyrimidine-based JAK2 inhibitor for the treatment of myeloid and lymphoid malignancies. Leukemia. 2011;25(11):1751–9.
Komrokji RS, Wadleigh M, Seymour JF, Roberts AW, To LB, Zhu HJ, Mesa RA. Results of a Phase 2 study of pacritinib (SB1518), a novel oral JAK2 Inhibitor, in patients with primary, post-polycythemia vera, and post-essential thrombocythemia myelofibrosis. Blood. 2011;118:282 (ASH Annual Meeting Abstracts 2011).
Verstovsek S, Mesa RA, Rhoades SK, Giles JLK, Pitou C, Jones E, Walgren RA, Prchal JT. Phase I study of the JAK2 V617F inhibitor, LY2784544, in patients with myelofibrosis (MF), polycythemia vera (PV), and essential thrombocythemia (ET). Blood. 2011;118:2814 (ASH Annual Meeting Abstracts 2011).
Nakaya Y, et al. Efficacy of NS-018, a potent and selective JAK2/Src inhibitor, in primary cells and mouse models of myeloproliferative neoplasms. Blood Cancer J. 2011;1(7):22.
Hedvat M, et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16(6):487–97.
Purandare AV, et al. Characterization of BMS-911543, a functionally selective small-molecule inhibitor of JAK2. Leukemia. 2012;26(2):280–8.
Andraos R, et al. Modulation of activation-loop phosphorylation by JAK inhibitors is binding mode dependent. Cancer Discov. 2012;2(6):512–23.
Krempler A, et al. Generation of a conditional knockout allele for the Janus kinase 2 (Jak2) gene in mice. Genesis. 2004;40(1):52–7.
Mullally A, et al. Physiological Jak2V617F expression causes a lethal myeloproliferative Neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell. 2010;17(6):584–96.
Koppikar P, et al. Efficacy of the JAK2 inhibitor INCB16562 in a murine model of MPLW515L-induced thrombocytosis and myelofibrosis. Blood. 2010;115(14):2919–27.
Deshpande A, et al. Kinase domain mutations confer resistance to novel inhibitors targeting JAK2V617F in myeloproliferative neoplasms. Leukemia. 2012;26(4):708–15.
Marit MR, et al. Random mutagenesis reveals residues of JAK2 critical in evading inhibition by a tyrosine kinase inhibitor. PLoS ONE. 2012;7(8):e43437.
Weigert O, et al. Genetic resistance to JAK2 enzymatic inhibitors is overcome by HSP90 inhibition. J Exp Med. 2012;209(2):259–73.
Lucet IS, et al. The structural basis of Janus kinase 2 inhibition by a potent and specific pan-Janus kinase inhibitor. Blood. 2006;107(1):176–83.
Azam M, Latek RR, Daley GQ. Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell. 2003;112(6):831–43.
Koppikar P, et al. Heterodimeric JAK–STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature. 2012;489(7414):155–9.
Marubayashi S, et al. HSP90 is a therapeutic target in JAK2-dependent myeloproliferative neoplasms in mice and humans. J Clin Investig. 2010;120(10):3578–93.
Kalota, A., et al., Intrinsic resistance to JAK2 inhibition in myelofibrosis. Clin Cancer Res. 2013;19(7):1729-39.
Fiskus W, et al. Heat shock protein 90 inhibitor is synergistic with JAK2 inhibitor and overcomes resistance to JAK2-TKI in human myeloproliferative neoplasm cells. Clin Cancer Res. 2011;17(23):7347–58.
Mascarenhas J, Mercado A, Rodriguez A, Lu M, Kalvin C, Li X, Petersen B, Najfeld V, Goldberg JD, Hoffman R. Prolonged low dose therapy with a pan-deacetylase inhibitor, panobinostat (LBH589), in patients with myelofibrosis. Blood, 2011;118:794 (ASH Annual Meeting Abstracts 2011).
Rambaldi A, et al. A pilot study of the histone-deacetylase inhibitor givinostat in patients with JAK2V617F positive chronic myeloproliferative neoplasms. Br J Haematol. 2010;150(4):446–55.
Rambaldi A, Finazzi G, Vannucchi AM, Martinelli V, Rodeghiero F, Nobile F, Specchia G, Pogliani EM, Olimpieri OM, Fioritoni G, Musolino C, Saglio G, Sivera P, Barosi G, Tollo S Di, Barbui T. A Phase II study of the HDAC inhibitor givinostat in combination with hydroxyurea in patients with polycythemia vera resistant to hydroxyurea monotherapy. Blood. 2011;118:1748 (ASH Annual Meeting Abstracts 2011).
Wang Y, et al. Cotreatment with panobinostat and JAK2 inhibitor TG101209 attenuates JAK2V617F levels and signaling and exerts synergistic cytotoxic effects against human myeloproliferative neoplastic cells. Blood. 2009;114(24):5024–33.
Baffert F, Evrot E, Ebel N, Roelli C, Andraos R, Qian Z, Romanet V, Murakami M, Radimerski T. Improved efficacy upon combined JAK1/2 and pan-deacetylase inhibition using ruxolitinib (INC424) and panobinostat (LBH589) in preclinical mouse models of JAK2V617F-driven disease. Blood. 2011;118:798 (ASH Annual Meeting Abstracts 2011).
Guglielmelli P, et al. Safety and efficacy of everolimus, a mTOR inhibitor, as single agent in a Phase 1/2 study in patients with myelofibrosis. Blood. 2011;118(8):2069–76.
Fiskus W, et al. Dual PI3K/AKT/mTOR inhibitor BEZ235 synergistically enhances the activity of JAK2 inhibitor against cultured and primary human myeloproliferative neoplasm cells. Mol Cancer Ther. 2013. doi:10.1158/1535-7163.MCT-12-0862.
Suryani S, Sia KCS, Bracken LC, Hernan EK, Kurmasheva R, Houghton PJ, Smith MA, Lock RB. Dual Inhibition of JAK/STAT and MAPK pathways results in synergistic cell killing of JAK-mutated pediatric acute lymphoblastic leukemia. Blood. 2012;120:3562 (ASH Annual Meeting Abstracts 2012).
Fiskus W, Manepalli RR, Balusu R, Bhalla KN. Synergistic activity of combinations of JAK2 kinase inhibitor with PI3K/mTOR, MEK or PIM kinase inhibitor against human myeloproliferative neoplasm cells expressing JAK2V617F. Blood. 2010;116:798 (ASH Annual Meeting Abstracts 2010).
Zhao C, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458(7239):776–9.
Dierks C, et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on hedgehog pathway activation. Cancer Cell. 2008;14(3):238–49.
Heidel Florian H, et al. Genetic and pharmacologic inhibition of β-catenin targets imatinib-resistant leukemia stem cells in CML. Cell Stem Cell. 2012;10(4):412–24.
Heidel F, Mar B, Armstrong S. Self-renewal related signaling in myeloid leukemia stem cells. Int J Hematol. 2011;94(2):109–17.
Jamieson C, Cortes JE, Oehler V, Baccarani M, Kantarjian HM, Papayannidis C, Rice KN, Zhang X, Shaik N, Courtney R, Levin WJ, Martinelli G. Phase 1 dose-escalation study of PF-04449913, an oral hedgehog (Hh) inhibitor, in patients with select hematologic malignancies. Blood. 2011;118:424 (ASH Annual Meeting Abstracts 2011).
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Bhagwat, N., Levine, R.L. & Koppikar, P. Sensitivity and resistance of JAK2 inhibitors to myeloproliferative neoplasms. Int J Hematol 97, 695–702 (2013). https://doi.org/10.1007/s12185-013-1353-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-013-1353-5