
Abstract
Multiple myeloma (MM) is characterized by the clonal expansion of malignant plasma cells. As in other cancers, MM plasma cells are thought to be derived from MM-initiating cells, although these remain unidentified. MM patients harbor phenotypic CD19+ B cells expressing the immunoglobulin gene sequence and the idiotype unique to the individual myeloma clone. Some previous studies have reported that CD19+ clonotypic B cells can serve as MM-initiating cells. However, we and another group have recently showed that CD19+ B cells from many MM patients do not reconstitute MM disease upon transplantation into NOD/SCID IL2Rγc−/− mice. In the SCID-rab and SCID-hu models, which enable engraftment of human MM in vivo, CD19−CD38++ plasma cells engrafted and rapidly propagated MM, while engraftment of CD19+ B cells was not detected. Both CD138− and CD138+ plasma cells have the potential to propagate MM clones in vivo in the absence of CD19+ B cells. Distinct from acute myeloid leukemia-initiating cells, which are derived from undifferentiated stem or progenitor cells, MM-initiating cells are derived from plasma cells, which are terminally differentiated cells. An improved understanding of how the bone marrow microenvironment supports MM-initiating plasma cells, which can initiate MM disease in the SCID-hu (or rab) model, is thus now essential.
Similar content being viewed by others


CD19+ clonotypic B cells in MM patients
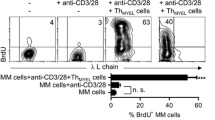
Multiple myeloma (MM) is characterized by the clonal expansion of malignant plasma cells [1–3]. The immunoglobulin gene sequences in MM plasma cells are somatically hyper-mutated and remain constant throughout the clinical course, suggesting that the disease arises from a post-germinal center B cell or a more differentiated cell [4–6] (Fig. 1). Previous studies have found that MM patients harbor phenotypic B cells expressing the immunoglobulin gene sequence and the idiotype unique to the individual myeloma clone [7–10]. These findings imply that clonotypic B cells may be involved in the disease process but offer no definitive proof that B cells in fact correspond to the proliferating tumor compartment. We examined the expression of immunoglobulin light chain κ and λ in the CD19+ B cells and CD19−CD38++ plasma cells from MM patients, and found highly biased expression of κ or λ chain in CD19+ B cells in some patients, suggesting that clonal CD19+ B cells expanded in these patients. Morphological abnormality of CD19+ B cells were also observed in some patients (Fig. 2). It is thus no doubt that clonotypic B cells can be found in MM patients, however, it has been unclear whether clonotypic B cells are MM-initiating cells or pre-malignant cells, which need additional oncogenic hits to become fully transformed MM-initiating cells.

MM-initiating cells originate from post-germinal center B cells. Differentiation steps from hematopoietic stem cells to plasma cells are shown. VDJ recombination and somatic hypermutations produce the varieties in immunoglobulin sequences. IgH VDJ sequences of MM plasma cells contain somatic hypermutation but no clonal variation, indicating that post-germinal center B cells are the origin of MM-initiating cells

CD19+ B cells from MM patients are not normal. FACS analysis of BM cells from MM patients. A representative analysis is shown. IgLk/l expression and May–Giemsa staining of CD19+ B cells and CD19−CD38++ plasma cells are shown
MM-initiating potentials of CD19+ clonotypic B cells could not be detected by xeno-transplant assay using NOD/SCIDXIL2Rγ−/− mice in many MM patients
CD19+ B cells isolated from MM patients could reportedly generate MM disease upon transplantation into NOD/SCID mice in a few MM patients [11–14], indicating that clonotypic CD19+ B cells served as MM progenitor cells in these patients. These results suggested that CD19+ clonotypic B cells are important therapeutic targets in MM therapy. However, B cell depletion by means of rituximab in MM patients was not clinically effective in most cases, at least for short periods [15]. It is therefore still unclear whether CD19+ or CD20+ clonogenic MM progenitor cells are responsible for disease progression and maintenance.
We transplanted purified CD19+ B cells from peripheral blood (PB) or bone marrow (BM) of MM patients into immunodeficient mice. CD19+ B cells were FACS-sorted from PB or BM cells from MM patients and transplanted directly into BM of NOD/Shi-scid,IL-2Rγnull (NOG) mice or intravenously to new born pups of NOG mice [16]. Engraftment of human MM cells was monitored by measuring human immunoglobulin light chain (IgL) κ and λ in serum of the recipient mice, but no human IgL was detected at any time. We also analyzed BM of the recipient mice 12–20 weeks after transplant, but no human CD19+ or CD38++ cells were detected (Fig. 3a). CD19−CD38++ plasma cells from the MM BM samples were also transplanted into NOG mice, but did not engraft. In contrast, robust engraftment of human cells was observed upon transplantation of cord blood-derived CD34+ cells (Fig. 3a). In some experiments, CD3−CD34−CD138− cells were transplanted into BM of NOG mice, but did not engraft. Finally, CD3-depleted BM cells from MM patients were transplanted intravenously into a newborn NOG mouse. When the BM cells were analyzed 12 weeks after transplant, significant engraftment of human CD45+ cells was observed because CD3− BM cells contained many CD34+ hematopoietic stem and progenitor cells. Small numbers of human CD38++CD138+ plasma cells were also detected, but analysis of their IgL κ and λ expression showed that they were not clonal MM plasma cells (Fig. 3b). This result suggests that normal human hematopoietic cells, but not MM cells engrafted in the recipient mice. Kim and Weissman [17] have recently reported that the similar results.

CD19+ B cells from MM samples did engraft and initiate MM disease in NOG mice. a FACS analysis of BM cells of NOG mice transplanted with CD19+ B cells from the patient’s BM. Corresponding data for an NOG mouse transplanted with cord blood-derived CD34+ cells is shown for reference. Analyses were performed 12 weeks post-transplant. b FACS analysis findings of NOG mice transplanted with CD3− BM cells from a MM patient sample 12 weeks after transplant. Expression of cytoplasmic IgLκ and λ in CD38++CD138+ cells was also analyzed to determine whether they were clonal MM cells
Taken together, the presence of CD19+ MM-initiating cells could not be proven by xeno-transplant assay using NOG mice as recipients in many of MM patients, although it is certain that there are small numbers of MM cases in which CD19+ B cells has MM-initiating potential as previously reported.
CD19−CD38++ MM plasma cells, but not CD19+ B cells, can initiate and maintain MM disease in the SCID-hu/rab system
SCID-hu or SCID-rab model has been used for reconstitution of MM disease in mice [18, 19]. In Japan, only SCID-rab model is permitted to perform. A human or rabbit bone fragment is inserted under the skin of a SCID mouse more than 4 weeks before transplantation of MM cells. Samples from MM patients are injected into the human or rabbit bone, and the engraftment and expansion of MM cells can be detected by measuring human immunoglobulin light chain (IgL) κ and λ in serum of the recipient mouse. We first transplanted whole BM cells from MM patients and engraftment of MM cells was monitored by measuring human IgLκ and λ in serum of the recipient mice. Engraftment and expansion of MM cells were observed in 5 out of 12 cases. Rabbit BM was analyzed 12 weeks or more after transplant to determine whether engraftment of not only MM plasma cells but also CD19+ B cells had taken place. Robust engraftment of human CD38++ MM plasma cells expressing the monotypic immunoglobulin light chain and containing both CD138− and CD138+ cells was detected in the rabbit BM, but no human CD19+ B cells were detected. These results indicate that CD19−CD38++ plasma cells could engraft and expand at least for several months without engraftment of CD19+ B cells. The same results have been recently reported using SCID-hu system by Kim and Weissman [17].
It was reported that clonogenic MM progenitor cells could be found in the CD138− population in MM patients. We thus transplanted purified CD138− or CD138+ BM cells into SCID-rab mice to test whether proliferating cell compartments were present in the CD138− population. Transplants with 9 MM samples were performed and in 3 of the samples, rapid increase of either human IgLκ or λ was observed in serum of the mice transplanted with CD138− cells. In the rabbit BM, CD38++ MM plasma cells including CD138− and CD138+ cells, but not CD19+ cells, were detected (Fig. 4). In 4 of 8 cases, CD138+ plasma cells also engrafted and expanded, although more slowly than CD138− BM cells. Rabbit bone engrafted with CD138+ cells contained both CD38++CD138− and CD38++CD138+ plasma cells (Fig. 4). Importantly, these BM cells, which do not contain CD19+ B cells, could be secondary transplanted to another SCID-rab recipient and propagate MM disease (Fig. 4). These results indicate that both CD38++CD138− and CD38++CD138− plasma cells, but not CD19+ B cells, contained MM-initiating cells, which have the potential to propagate and maintain MM clones, at least for several months, in the absence of clonotypic B cells. In some patients, highly proliferative MM progenitor cells were present in the CD138-negative fraction, but those cells were MM plasma cells, not B cells (Fig. 4). CD138− MM plasma cells may be enriched with immature and proliferative plasma cells. In consistent with this, Kawano et al. [20] have recently reported that MM cells expressing low levels of CD138 have an immature phenotype and resistance against lenalidomide.

Both CD138− and CD138+ plasma cells, but not CD19+ B cells, could engraft and propagate MM clones in the SCID-rab model. Transplantation of purified CD138−CD34− or CD138+ cells from MM BM cells into SCID-rab recipients. Concentration of human IgL in serum at 12 weeks post-transplant and the results of analysis of BM cells at 12 weeks (CD138−) or 24 weeks (CD138+) are shown
Interestingly, substantial numbers of CD138−CD38++ plasma cells were detected in the SCID-rab mice transplanted with CD138+ plasma cells. This suggests that CD138 expression on MM plasma cells may be reversible. The significance of CD138 expression on clonogenic MM cells thus needs to be carefully interpreted. It was reported that interaction with bone marrow stroma cells induced expression of CD138 in MM plasma cells [21], indicating that changes in CD138 expression depend on the microenvironment. Jakubikova et al. [22] have recently reported that clonogenic side populations in MM cells were not enriched in the CD138low/+ but not in the CD138− population, although it is not clear whether clonogenic side populations in primary MM cells are also enriched in CD138low/+ cells.

MM-initiating cells are present in the MM plasma cells. The summary of our studies about MM-initiating cells are shown. In most MM patients, clonotyopic CD19+ B cells are likely to be pre-MM-initiating cells, and fully transformed MM-initiating cells are present in the plasma cell populations. To eradicate MM-initiating cells in plasma cells, monoclonal antibodies against plasma cells are one promising strategy, and CD48 is a new candidate target for it
Taken together, our findings show that MM-initiating cells are present in plasma cells in most MM patients. Both CD138− and CD138+ plasma cells have potential to propagate and maintain MM clones for at least several months without the need for CD19+ clonotypic B cells (Fig. 5).
Future prospects about studies about MM-initiating cells or MM stem cells
It is still unclear whether only a subset of plasma cells has MM-initiating potential or many plasma cells have it. It is well known that the survival of MM plasma cells is highly dependent on microenvironment [23, 24]. Thus, it may be possible that many plasma cells, even if not all, can acquire MM-initiating potential by being at an appropriate location namely “niche”. It is undoubtedly important to know more about the niche of MM plasma cells or MM-initiating cells. For long years, many researchers have been used “bone marrow stroma cell”, which are the mixture of non-hematopoietic cells in BM, as supporting cells for MM plasma cells. However, there has been only a little information about niche cells for MM plasma cells. Recently, Iriuchishima et al. [25] have reported interesting findings about niche for MM cells. By transplanting GFP-expressing human MM cell line into NOG mice, they found that transplanted MM cells preferentially engrafted at the metaphyseal region of the BM endosteum and formed a complex with osteoblasts and osteoclasts, suggesting that MM plasma cells have the character similar to quiescent hematopoietic stem cells (HSCs). Another interesting finding in this report was that a subpopulation of MM cells expressed VE-cadherin after transplantation and formed endothelial-like structures in the BM, and VE-cadherin(+) MM cells were chemo-resistant, hypoxic, and HIF-2alpha-positive compared to the VE-cadherin(−) population, suggesting the possibility of the existence of VE-cadherin(+) MM stem cells. Accumulating knowledge about stem cell niche obtained from HSC research [26] may be applied to studies of MM stem cells.
CD19+ B cells in a few MM patients generated MM disease upon transplantation into NOD/SCID mice [11–14], indicating that CD19+ B cells of these MM patients definitely contain MM progenitor cells. In our experiments [16] and those by Kim et al. [17], CD19+ B cells from MM patients never induced MM disease upon transplantation into NOG mice. However, this does not necessarily mean that CD19+ clonotypic B cells cannot be MM progenitor cells in those MM patients. It should be noted that there are many difficulties involved in the engraftment of human cells in xeno-graft models. For example, mouse IL6, which is one of the major growth factors for plasma cells, does not transduce its signals through human IL6 receptors, and probably other factors also lack inter-species cross-reactivity between human and mice. This means that MM progenitor cells can be detected in xeno-graft assays only when they can survive independently of human IL6 or other human factors. Thus, CD19+ B cells from advanced MM patients may be independent of several survival factors and effectively engraft and propagate MM disease upon transplant into immuno-deficient mice. In addition, signals from B cell receptors (BCRs) on clonotypic CD19+ B cells need to be taken into consideration. When BCRs of clonotypic CD19+ B cells show very high affinity to xeno-antigen in mice, they may be depleted and cannot survive in xeno-graft models. Thus, assuming that CD19+ MM progenitor cells are present, they can be detected in xeno-graft assays only when their BCRs are suitable for survival in mice.
While xeno-transplant system is hard to utilize in research for the stem cells of B cell malignancies for the reason as described above, it is an extremely excellent tool for the analysis of human hematopoietic stem cells. Numbers of excellent studies about normal or malignant hematopoietic stem cells have been done with xeno-transplantation to NOD/SCID or NOG mice. It is thus possible for us to analyze whether hematopoietic stem cells are normal in MM or other B cell malignancies. Kikushige and Akashi [27] have recently reported surprising results of the analysis of HSCs in CLL patients. They found that HSCs purified from patients with CLL displayed lymphoid-lineage gene priming and produced a high number of polyclonal B cell progenitors. Strikingly, their maturation into B cells was restricted always to mono- or oligo-clones with CLL-like phenotype in xenogeneic recipients. These results let us prompted us to perform the same analysis of HSCs from MM patients.
How to target MM-initiating cells in bone marrow
MM-initiating plasma cells should be considered important therapeutic targets, although we have not identified which plasma cells have MM-initiating potential. The Hedgehog signaling pathway was reported to be a promising candidate as a therapeutic target against clonogenic MM cells [28]. In addition, it is also important to understand the mechanisms involved in how the BM microenvironment supports clonogenic MM plasma cells and targets them. The Notch signaling pathway may be a good candidate for such a target [29].
In addition to these, therapy with monoclonal antibody against MM plasma cells should be a promising strategy [30]. Since the mechanisms of antibody drug to kill tumor cells are totally different from those of other drugs such as chemotherapeutic drugs, proteasome inhibitors, and IMIDs, combination of antibody drugs with these conventional therapies should be effective. In fact, the addition of rituximab to CHOP chemotherapy increased the cure rate of lymphoma patients. Results of clinical trials with anti-CS1 antibody is very promising [31], and thereby it is expected to be approved as the first antibody drug against MM. In addition, several other antibodies, for example anti-CD38 mAb [32], have been also proved to be effective in MM patients.
We recently identified CD48 as a novel therapeutic target that is highly expressed on MM plasma cells in almost all MM patients (Fig. 5) [33]. In 22 out of 24 MM patients, CD48 was expressed on more than 90 % of MM plasma cells at significantly higher levels than it was on normal lymphocytes and monocytes. An anti-CD48 mAb that was newly generated by us induced antibody-dependent cell-mediated cytotoxicity and marked complement-dependent cytotoxicity against not only MM cell lines but also primary MM plasma cells in vitro. Administration of the anti-CD48 mAb significantly inhibited tumor growth in SCID mice inoculated subcutaneously with MM cells. Furthermore, anti-CD48 mAb treatment inhibited growth of MM cells transplanted directly into BM of the mice. Finally and importantly, we demonstrated that the anti-CD48 mAb did not damage normal CD34+ hematopoietic stem/progenitor cells. It should be noted that the expression patterns of CD48 in human hematopoietic progenitor cells are largely different form murine ones. While CD48 is expressed on almost all of c-kit+Sca-1+lin− mouse HSCs except for CD150+ long term-HSCs, in most of human CD34+ hematopoietic stem cells do not express or expressed at very low levels of CD48. These results suggest that the anti-CD48 mAb has the potential to become an effective therapeutic mAb against MM.
Conclusion
Different from acute myeloid leukemia-initiating cells, which are derived from undifferentiated stem or progenitor cells, MM-initiating cells are derived from plasma cells, which are terminally differentiated cells. This suggests that strategies for targeting MM-initiating cells should be different from those for leukemia-stem cells. On the other hand, some reports suggested common characters between MM plasma cells and hematopoietic stem cells. Especially, both of them highly prefer BM microenvironment. We now need to know how BM microenvironment supports MM-initiating plasma cells, which can initiate MM disease in the SCID-hu (or rab) model.
References
Kyle RA, Rajkumar SV. Multiple myeloma. N Engl J Med. 2004;351:1860–73.
Kyle RA, Rajkumar SV. Multiple myeloma. Blood. 2008;111:2962–72.
Kapoor P, Rajkumar SV. Update on risk stratification and treatment of newly diagnosed multiple myeloma. Int J Hematol. 2011;94(4):310–20.
Bakkus MH, Heirman C, Van Riet I, Van Camp B, Thielemans K. Evidence that multiple myeloma Ig heavy chain VDJ genes contain somatic mutations but show no intraclonal variation. Blood. 1992;80:2326–35.
Vescio RA, Cao J, Hong CH, et al. Myeloma Ig heavy chain V region sequences reveal prior antigenic selection and marked somatic mutation but no intraclonal diversity. J Immunol. 1995;155:2487–97.
Sahota SS, Leo R, Hamblin TJ, Stevenson FK. Myeloma VL and VH gene sequences reveal a complementary imprint of antigen selection in tumor cells. Blood. 1997;89:219–26.
Pilarski LM, Jensen GS. Monoclonal circulating B cells in multiple myeloma. A continuously differentiating, possibly invasive, population as defined by expression of CD45 isoforms and adhesion molecules. Hematol Oncol Clin North Am. 1992;6:297–322.
Bergsagel PL, Smith AM, Szczepek A, Mant MJ, Belch AR, Pilarski LM. In multiple myeloma, clonotypic B lymphocytes are detectable among CD19+ peripheral blood cells expressing CD38, CD56, and monotypic Ig light chain. Blood. 1995;85:436–47.
Chen BJ, Epstein J. Circulating clonal lymphocytes in myeloma constitute a minor subpopulation of B cells. Blood. 1996;87:1972–6.
Rasmussen T, Jensen L, Johnsen HE. The CD19 compartment in myeloma includes a population of clonal cells persistent after high-dose treatment. Leuk Lymphoma. 2002;43:1075–7.
Pilarski LM, Seeberger K, Coupland RW, et al. Leukemic B cells clonally identical to myeloma plasma cells are myelomagenic in NOD/SCID mice. Exp Hematol. 2002;30:221–8.
Pilarski LM, Hipperson G, Seeberger K, Pruski E, Coupland RW, Belch AR. Myeloma progenitors in the blood of patients with aggressive or minimal disease: engraftment and self-renewal of primary human myeloma in the bone marrow of NOD SCID mice. Blood. 2000;95:1056–65.
Matsui W, Huff CA, Wang Q, et al. Characterization of clonogenic multiple myeloma cells. Blood. 2004;103:2332–6.
Matsui W, Wang Q, Barber JP, et al. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res. 2008;68:190–7.
Kapoor P, Greipp PT, Morice WG, Rajkumar SV, Witzig TE, Greipp PR. Anti-CD20 monoclonal antibody therapy in multiple myeloma. Br J Haematol. 2008;141:135–48.
Hosen N, Matsuoka Y, Kishida S, et al. CD138-negative clonogenic cells are plasma cells but not B cells in some multiple myeloma patients. Leukemia. 2012;26:2135–41.
Kim D, Park CY, Medeiros BC, Weissman IL. CD19(−)CD45(low/−)CD38(high)/CD138(+) plasma cells enrich for human tumorigenic myeloma cells. Leukemia. 2012;26(12):2530–7.
Yaccoby S, Barlogie B, Epstein J. Primary myeloma cells growing in SCID-hu mice: a model for studying the biology and treatment of myeloma and its manifestations. Blood. 1998;92:2908–13.
Yata K, Yaccoby S. The SCID-rab model: a novel in vivo system for primary human myeloma demonstrating growth of CD138-expressing malignant cells. Leukemia. 2004;18:1891–7.
Kawano Y, Fujiwara S, Wada N, et al. Multiple myeloma cells expressing low levels of CD138 have an immature phenotype and reduced sensitivity to lenalidomide. Int J Oncol. 2012;41(3):876–84.
Fuhler GM, Baanstra M, Chesik D, et al. Bone marrow stromal cell interaction reduces syndecan-1 expression and induces kinomic changes in myeloma cells. Exp Cell Res. 2010;316:1816–28.
Jakubikova J, Adamia S, Kost-Alimova M, et al. Lenalidomide targets clonogenic side population in multiple myeloma: pathophysiologic and clinical implications. Blood. 2011;117:4409–19.
Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer. 2007;7(8):585–98.
Abe M. Targeting the interplay between myeloma cells and the bone marrow microenvironment in myeloma. Int J Hematol. 2011;94(4):334–43.
Iriuchishima H, Takubo K, Miyakawa Y, et al. Neovascular niche for human myeloma cells in immunodeficient mouse bone. PLoS One. 2012;7(2):e30557.
Arai F, Yoshihara H, Hosokawa K, et al. Niche regulation of hematopoietic stem cells in the endosteum. Ann N Y Acad Sci. 2009;1176:36–46.
Kikushige Y, Ishikawa F, Miyamoto T, et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell. 2011;20(2):246–59.
Peacock CD, Wang Q, Gesell GS, et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci USA. 2007;104:4048–53.
Nefedova Y, Sullivan DM, Bolick SC, Dalton WS, Gabrilovich DI. Inhibition of Notch signaling induces apoptosis of myeloma cells and enhances sensitivity to chemotherapy. Blood. 2008;111:2220–9.
Hideshima T, Anderson KC. Novel therapies in MM: from the aspect of preclinical studies. Int J Hematol. 2011;94(4):344–54.
Benson DM Jr, Byrd JC. CS1-directed monoclonal antibody therapy for multiple myeloma. J Clin Oncol. 2012;30(16):2013–5.
de Weers M, Tai YT, van der Veer MS, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol. 2011;186(3):1840–8.
Hosen N, Ichihara H, Mugitani A, et al. CD48 as a novel molecular target for antibody therapy in multiple myeloma. Br J Haematol. 2011;156:213–24.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Hosen, N. Multiple myeloma-initiating cells. Int J Hematol 97, 306–312 (2013). https://doi.org/10.1007/s12185-013-1293-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-013-1293-0