
Abstract
NarE is a 16 kDa protein identified from Neisseria meningitidis, one of the bacterial pathogens responsible for meningitis. NarE belongs to the ADP-ribosyltransferase family and catalyses the transfer of ADP-ribose moieties to arginine residues in target protein acceptors. Many pathogenic bacteria utilize ADP-ribosylating toxins to modify and alter essential functions of eukaryotic cells. NarE was proposed to bind iron through a Fe–S center which is supposed to be implied in catalysis. We have produced and purified uniformly labeled 15N- and 15N/13C-NarE and assigned backbone and side-chain resonances using multidimensional heteronuclear NMR spectroscopy. These assignments provide the starting point for the three-dimensional structure determination of NarE and the characterization of the role of the Fe–S center in the catalytic mechanism.
Similar content being viewed by others


Biological context
Neisseria meningitidis is a gram-negative bacterium best known for its role in meningitis and septicemia. This human-specific pathogen is considered as a major cause of morbidity and mortality during childhood in industrialized countries and is responsible for epidemics in Africa and in Asia (Genco and Wetzler 2010). Like most bacterial pathogens, N. meningitidis synthesizes a number of toxic products believed to target and kill their host cells. Among these toxins, proteins that exert ADP-ribosylation activity represent a large family of potentially lethal enzymes able to modify or disrupt essential functions of eukaryotic cells (Moss and Vaughan 1988). ADP-ribosyltransferases catalyze the transfer of a single ADP-ribosyl group from β-nicotinamide adenine dinucleotide (NAD+) onto specific amino acid residues of host cell proteins with the simultaneous release of nicotinamide (Althaus and Richter 1987). Many bacterial pathogens use ADP-ribosylating enzymes to block protein synthesis or to alter signal transduction by inactivating key target proteins such as GTP-binding proteins (Locht and Keith 1986).
The putative ADP-ribosylating NarE protein from N. meningitidis was recently identified on the basis of a profile-based computational approach (Masignani et al. 2003). NarE shares structural features with toxins from V. cholerae and E. coli, and, like cholera toxin and heat-labile enterotoxins, retains the capacity to ADP-ribosylate arginine and to hydrolyze NAD in ADP-ribose and nicotinamide. Another feature of NarE is its ability to bind iron through an iron–sulfur center (Fe–S) (Del Vecchio et al. 2009). Interestingly, site-directed mutagenesis and enzymatic assays showed that correct assembly of the iron-binding site is essential for transferase but not for hydrolase activity (Del Vecchio et al. 2009), suggesting for the first time an implication of a Fe–S cluster in catalysis within the ADP-ribosyltransferase family. We here report the 1H, 15N and 13C assignments of NarE protein from N. meningitidis as a preliminary step towards characterizing its three-dimensional structure and elucidating the role of the iron–sulfur center in the catalytic mechanism.
Methods and experiments
The NarE gene was PCR amplified from the chromosomal DNA of N. meningitidis MC58 strain and cloned into a pET21b+ plasmid (Novagen) under the control of the T7 promoter. The resulting expression vector encodes a 17 kDa fusion protein that contains the wild-type NarE sequence (145 residues) followed by a 8-residue C-terminal histidine-tag (LEHHHHHH) used for purification purposes. The pET21b+ plasmid was transformed into an Escherichia coli BL21 (DE3) expression strain. Recombinant protein was prepared from cells grown to an OD600 of 0.5–0.6 at 37°C in a bacterial culture supplemented by 100 μg/ml ampicillin. Protein expression was then induced by addition of 1 mM isopropyl-1-thio-β-d-galactopyranoside (IPTG), and the culture was further grown for 3 h at 37°C. The cells were harvested by centrifugation (8,000×g for 30 min), resuspended in 50 mM phosphate buffer (pH 8.0) containing 300 mM NaCl, and lysed with a French press cell (SLM, Aminco). The soluble fraction enriched with the NarE protein was separated from the inclusion bodies by centrifugation at 39,200×g for 45 min, and loaded on a nickel affinity chromatography column (GE Healthcare). The protein was purified using IMAC standard protocols and an imidazole concentration of 125 mM was required to elute recombinant NarE. A second step of purification was performed using anion exchange QHP chromatography (GE Healthcare). Fractions containing the target protein were collected at a NaCl concentration of 300 mM and finally concentrated by ultrafiltration using a 3-kDa cutoff membrane (Millipore).
Expression of uniformly labeled 15N and 15N/13C proteins was carried out by growing BL21 strains in ISOGRO-15N and ISOGRO-15N, 13C rich media (Sigma), respectively. After buffer exchange and concentration, the resulting NMR samples contained approximately 0.6 mM NarE protein in 25 mM phosphate buffer (pH 7.5), 75 mM NaCl, 1 mM PMSF, a protease inhibitor cocktail (Boehringer), 1 mM NaN3, and either 90% H2O/10% D2O or 100% D2O. Trimethylsilyl-[2,2,3,3-2H4] propionate (TSP) was added as an internal 1H chemical shift reference. 13C and 15N chemical shifts were referenced indirectly to TSP, using the absolute frequency ratios (Wishart et al. 1995). NMR experiments were performed at 27°C on a Bruker Avance-II 750 MHz equipped with a triple-resonance (1H, 15N, 13C) probe. Spectra recorded for sequential backbone assignment were as follows: 2D 15N–1H HSQC, 3D HNCO, 3D HN(CA)CO, 3D HNCA, 3D CBCA(CO)NH, 3D CBCANH, and 3D HBHA(CO)NH. Aliphatic and aromatic 1H and 13C side chain assignments were obtained using 3D HCCH-TOCSY, 3D HCCH-COSY, and 3D (H)CC(CO)NH experiments. All NMR data were processed with the NMRPipe software (Delaglio et al. 1995) and analysed with Sparky (Goddard and Kneller).
Extent of assignments and data deposition
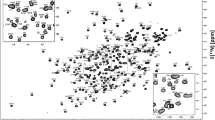
Figure 1 shows the assigned 1H–15N HSQC spectrum recorded on the 15N-labeled protein sample. In this spectrum most of the cross-peaks are well-dispersed, indicating that recombinant NarE is well-folded in the NMR aqueous buffer. All the NH backbone resonances were assigned except for the N-terminal residue M1 and for S49 for which the absence of a cross-peak may be due to loop mobility or conformational exchange as this amino acid follows a proline in the NarE sequence. The lack of 13C assignments for residue P48 due to line broadening confirms the presence of dynamics in this region of the protein. Interestingly, cysteines C67 and C128, which could be shown to be crucial for the coordination of iron (Del Vecchio et al. 2009), have a C β chemical shift value higher than 40 ppm, suggesting the formation of an intramolecular disulfide bridge between them in the absence of a reducing agent. Since the used protein samples were colourless and line broadening of NMR signals due to paramagnetic effects of iron-III could not be observed, the presented resonance assignment likely refers to the protein’s apo-form.

Assigned 15N–1H HSQC spectrum of the 15N-labeled NarE protein acquired at 750 MHz and 27°C. Amide side-chain resonances are connected by solid lines
Overall, all residues corresponding to NarE were identified: the backbone assignment is complete for 1Hα and reaches 98% for 1HN, 15N, 13Cα and 13CO nuclei. 88% of the aliphatic side chain resonances were assigned and 94% of the aromatic 1H and 13C resonances were identified including all Phe and Tyr residues. The assignment of NH2 side-chain resonances from asparagines and glutamines were also complete except for N145. The chemical shifts have been deposited into the BioMagResBank (http://www.bmrb.wisc.edu) under accession number 16737. The CSI (Wishart and Sykes 1994) and TALOS+ (Shen et al. 2009) programs were used to evaluate NarE secondary structure from 1Hα, 13Cα, 13CO and 13Cβ chemical shifts. As shown in Fig. 2, both methods are in agreement with the presence of two α-helices (residues 11–15, and 76–81) and six β-strands (residues 4–9, 68–72, 88–96, 105–111, 118–125 and 136–143).

Secondary structure prediction for the NarE sequence as obtained from TALOS+ and consensus chemical shift index (CSI) programs. The regions identified to adopt α-helical and β-strand secondary structures are named α and β, respectively
References
Althaus FR, Richter C (1987) ADP-ribosylation of proteins. Enzymology and biological significance. Mol Biol Biochem Biophys 37:1–237
Del Vecchio M, Pogni R, Baratto MC, Nobbs A, Rappuoli R, Pizza M, Balducci E (2009) Identification of an iron–sulfur cluster that modulates the enzymatic activity in NarE, a Neisseria meningitides ADP-ribosyltransferase. J Biol Chem 284(48):33040–33047
Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6:277–293
Genco C, Wetzler L (eds) (2010) Neisseria: molecular mechanisms of pathogenesis. Caister Academic Press, Norwich, UK
Goddard TD, Kneller DG SPARKY 3. University of California, San Francisco. http://www.cgl.ucsf.edu/home/sparky/
Locht C, Keith JM (1986) Pertussis toxin gene: nucleotide sequence and genetic organization. Science 232:1258–1264
Masignani V, Balducci E, Di Marcello F, Savino S, Serruto D, Veggi D, Bambini S, Scarselli M, Arico B, Comanducci M, Adu-Bobie J, Giuliani M, Rappuoli R, Pizza M (2003) NarE: a novel ADP-ribosyltransferase from Neisseria meningitidis. Mol Microbiol 50(3):1055–1067
Moss J, Vaughan M (1988) ADP-ribosylation of guanyl nucleotide-binding regulatory proteins by bacterial toxins. Adv Enzymol 61:303–379
Shen Y, Delaglio F, Cornilescu G, Bax A (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR 44:213–223
Wishart DS, Sykes BD (1994) The 13C chemical shift index. A simple method for the identification of protein secondary structure using 13C chemical shift data. J Biomol NMR 4:171–180
Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes BD (1995) 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J Biomol NMR 6:135–140
Acknowledgments
This work was supported by the Sixth Research Framework Programme of the European Community, FP6-STREP project ‘‘BacAbs’’, grant number LSHB-CT-2006-037325.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Carlier, L., Koehler, C., Veggi, D. et al. NMR resonance assignments of NarE, a putative ADP-ribosylating toxin from Neisseria meningitidis . Biomol NMR Assign 5, 35–38 (2011). https://doi.org/10.1007/s12104-010-9261-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12104-010-9261-6