
Abstract
Background
Sarcoidosis is a systemic inflammatory disease that is characterized by non-caseating epithelioid-cell granulomas upon histology. However, similar histological findings may also be seen with certain infections. Thus, differentiation from infection is pivotal to ensure appropriate treatment. Here, we present a case of a disseminated infection with Mycobacterium genavense owing to an interleukin 12 receptor subunit beta 1 (IL-12Rβ1) associated immunodeficiency in a previously healthy female who was initially misdiagnosed with sarcoidosis. M. genavense is a nontuberculous mycobacterium which can cause lymphadenopathy, gastrointestinal and bone marrow infiltration in immunocompromised patients. With this case report we aim to highlight that an infection with M. genavense on the ground of a genetic defect of mycobacterial immune control may represent a rare differential diagnosis of sarcoidosis.
Case presentation
A 31-year-old female was referred to our hospital with progressive lymphadenopathy, hepatosplenomegaly, pancytopenia and systemic inflammation. She had previously been evaluated for generalized lymphadenopathy in another hospital. At that time, lymph node biopsies had revealed sarcoid-like lesions and a systemic corticosteroid treatment was initiated based on a putative diagnosis of sarcoidosis. When her condition worsened, she was transferred to our university clinic, where the diagnosis of disseminated M. genavense infection owing to an inborn interferonopathy was made. Her family history revealed that her brother had also suffered from IL-12Rβ1 deficiency and had died from a systemic infection with M. genavense at the age of 21. The patient received antimycobacterial treatment combined with subcutaneous type I interferon, which eventually led to a gradual improvement over the next months.
Conclusions
Differentiating between sarcoidosis and sarcoid-like lesions secondary to infections may be challenging, especially when pathogens are difficult to detect or not expected in an apparently immunocompetent patient. Patients with IL-12Rβ1-associated immunodeficiency may be asymptomatic until adulthood, and disseminated M. genavense infection on the grounds of an IL-12Rβ1-associated immunodeficiency may represent a rare differential diagnosis of sarcoidosis.
Similar content being viewed by others


Background
Sarcoidosis is an inflammatory multisystemic disease, and the diagnosis is established when clinical and radiological findings are compatible with the disease, and non-caseating epithelioid-cell granulomas are histologically described [1, 2]. However, the latter must be differentiated from sarcoid-like lesions secondary to infections, drugs, malignancy or other causes [1, 2]. Differentiation from infection can be challenging, especially when pathogens are rare and difficult to detect or not expected in an apparently immunocompetent patient.
Here, we present a case of a disseminated infection with the opportunistic pathogen Mycobacterium genavense owing to an interleukin 12 receptor subunit beta 1 (IL-12Rβ1) associated immunodeficiency in a previously healthy female who was initially misdiagnosed with sarcoidosis.
IL-12Rβ1 deficiency is an autosomal recessive, hereditary immunodeficiency that is part of a complex of genetic disorders termed Mendelian susceptibility to mycobacterial diseases (MSMD) [3]. MSMD comprises a number of rare genetic defects that affect the interaction between macrophages and T-cells and interfere with IFN-γ production [3]. Affected patients are susceptible to recurrent and disseminated infections with mycobacteria and other intracellular pathogens [3]. IL-12Rβ1 deficiency is the most common defect, and to date, mutations in 18 different genes associated with MSMD have been documented [3,4,5]. Patients with immunodeficiency caused by mutations in the IL-12Rβ1 gene often become symptomatic with infections in childhood. However, they may also remain asymptomatic until adulthood as the clinical phenotype is highly variable [3, 5].
Disseminated infections with M. genavense were first described in patients with human immunodeficiency virus (HIV), but have also been reported in non-HIV immunocompromised patients [6, 7]. Mahmood et al [7]. summarized 46 cases of M. genavense infections in non-HIV immunocompromised hosts, and found that 14% were associated with immunosuppressive treatment for sarcoidosis. With this case report we aim to highlight that an infection with M. genavense on the ground of a genetic defect of mycobacterial immune control may represent a rare but important differential diagnosis of sarcoidosis.
Case presentation
A 31-year-old female was referred to our tertiary care hospital with progressive pancytopenia, hepatosplenomegaly and systemic inflammation.
Twelve months prior to admission, she had been evaluated for right lower quadrant abdominal pain in another hospital. At that time, the diagnostic workup revealed terminal ileitis and a generalized lymphadenopathy. The biopsy of a mesenteric and a retroperitoneal lymph node showed granulomatous / sarcoid-like lesions upon histological evaluation. Based on the suspected diagnosis of sarcoidosis, a therapy with 0.5 mg Prednisolone per kilogram of body weight was initiated.
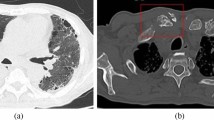
Over the following months, the patient’s condition gradually worsened, and she was transferred to our hospital for further evaluation. On admission, she complained of recurrent fever, night sweats and abdominal pain with predominance in the upper left quadrant. Upon physical examination, the patient was found to have sinus tachycardia and left upper abdominal quadrant tenderness. Blood pressure and oxygen saturation were within normal limits. Laboratory results showed new pancytopenia (leukocytes 1.8 G/l [reference range 4.0–10.0 G/l], hemoglobin 8.5 g/dl [reference range 12.0–15.7 g/dl], thrombocytes 72 G/l [reference range 150–380 G/l]) and systemic inflammation with a C-reactive protein of 14 mg/dl (reference range < 0.5 mg/dl), and an elevated erythrocyte sedimentation rate of 107 mm/h (reference range < 20 mm/h). Renal function, liver enzymes, and lactate dehydrogenase were within normal ranges, chest X ray and urine analysis showed no appreciable disease. 18FDG-PET/CT revealed progressive generalized and hypermetabolic lymphadenopathy (especially of mesenteric and retroperitoneal lymph nodes), a pronounced hypermetabolic splenomegaly, enhanced tracer uptake in the small intestine, and diffuse bone marrow hypermetabolism (Fig. 1). Serological or molecular analyses for human immunodeficiency virus, Epstein–Barr virus, cytomegalovirus, human herpesvirus 8, Salmonella typhi, Leishmania spp., Francisella tularensis and Yersinia enterocolitica were negative. Autoimmune testing, including serum ACE and sIL2R, was negative.

18FDG-PET analysis shows hypermetabolic lymphadenopathy and splenomegaly, together with enhanced tracer uptake in the small intestine, as well as a hypermetabolic bone marrow
The patient was of Turkish descent, and was born and raised in Germany. She had no premedication or allergies at the time of first presentation, and had not suffered from any serious acute or recurrent infections in the past. Importantly, examination of the patient’s family history revealed that her brother had died from a systemic infection with M. genavense at the age of 21, with an underlying IL-12Rβ1 deficiency. Her parents were both healthy and she had no other siblings. Neither the parents nor the patient had been genetically tested until the time of admission to our hospital.
At the time of the patient`s first presentation one year prior to the diagnosis of an IL-12Rβ1 deficiency, biopsy specimen of terminal ileum and lymph nodes were sent to two independent pathology laboratories. Ziehl–Neelsen staining was negative in all specimen. In one laboratory real-time PCR for Mycobacterium spp. (M. tuberculosis, M. bovis, M. bovis BCG, M. africanum, M. microti, M. canetti, M. pinipedii; Amplisens® MTC-FRT PCR Kit) was negative, while an independent laboratory reported a positive PCR result for atypical mycobacteria (LCD-Array Kit, Chipron). However, it did not provide further species information, as the additional testing of a predefined panel of nontuberculous mycobacteria (M. avium complex, M. kansasii, M. xenopi, M. abscessus, M. gordonae, M. peregrinum, M. szulgai, M. haemophilus, M. marinum/ulcerans, M. simiae and M. smegmatis) was negative. In our workup, Ziehl–Neelsen staining of bone marrow revealed massive infiltration with acid-fast bacilli (Fig. 2), and mycobacterial histiocytosis with displacement of hematopoietic cells was described. Pan-bacterial PCR of a bone marrow sample unveiled M. genavense, which was later confirmed by mycobacterial culture. Attempts to culture the pathogen from blood samples were not successful. With the diagnosis of disseminated infection with M. genavense and the positive family history, genetic investigations were initiated. These identified a homozygous pathogenic variant p(Cys198Arg) (c.592T > C) in the IL12RB1 gene (NM_005535, rs121434495), previously reported in attenuated IL-12Rβ1-associated immunodeficiency [8].

Ziehl–Neelsen staining of a bone marrow sample with numerous acid-fast bacilli and a dramatically reduced hematopoiesis
An antimycobacterial treatment with rifampin 600 mg q.d., ethambutol 1600 mg q.d. and clarithromycin 500 mg b.i.d. was initiated. After three weeks of treatment the patient developed erythema multiforme, and antimycobacterial treatment was switched to azithromycin 500 mg q.d., moxifloxacin 400 mg q.d., rifabutin 300 mg q.d., and intravenous amikacin 1000 mg q.d. The antimycobacterial treatment was combined with subcutaneous IFN-γ at 50 µg/kg body weight three times a week. The patient improved gradually over the next months. Amikacin was administered for 24 weeks and then discontinued due to tinnitus without hearing impairment upon tone audiogram. The treatment consisting of azithromycin, moxifloxacin, rifabutin and IFN-γ is ongoing at the time of submission.
Discussion and conclusions
M. genavense is a rare environmental mycobacterium that may cause opportunistic infections in immunocompromised patients, and it was first described in patients with HIV in the early 1990s [6]. Patients typically present with fever, night sweats, weight loss, hepatosplenomegaly, (especially mesenteric) lymphadenopathy, gastrointestinal symptoms, and high immune activation [7]. M. genavense is difficult to detect as it needs special conditions for cultivation and does not grow in standard mycobacterial media [6]. Although PCR has a high sensitivity, M. genavense is usually not part of PCR panels that are used for the detection of tuberculous and nontuberculous mycobacteria in routine clinical settings. Ziehl–Neelsen staining, on the other hand has a significantly lower sensitivity because of a detection limit of approximately 5,000–10,000 bacteria per ml [9]. In addition, M. genavense is a slow growing pathogen with low bacterial concentration, thus Ziehl–Neelsen staining may be false negative. Interferon-γ Release Assays are not adequate for the detection of nontuberculous mycobacteria and are not reliable in patients with IL-12Rβ1 deficiency, as an intact IL12/IFN-γ axis is needed [10]. In the here presented case, the diagnosis was established by bone marrow aspiration and subsequent pan-bacterial PCR.
M. genavense has been suggested to have a ubiquitous distribution as it was identified in tap water, the intestinal tract of immunocompetent humans and animals [11,12,13,14]. An intestinal/faecal-oral transmission has been theorized [15], however no human-to-human transmission has been reported so far. As the patient`s brother died of the same mycobacterial infection, infection from the same environmental exposure seems most likely, although a direct transmission cannot be fully excluded. The time of infection could not be determined.
Mycobacterial antigens have been discussed as trigger for sarcoidosis [16]. However, sarcoidosis secondary to an exposure to M. genavense seems unlikely in this case, as no improvement was observed upon previous corticosteroid therapy, rather did the patient deteriorate.
Patients with MSMD are at risk of recurrent and disseminated infections with mycobacteria as well as Salmonella, but also other intracellular pathogens [3]. This is due to the fact that IFN-γ and its inducible genes, such as TNF, IL-12 or iNOS are central for mounting an efficient host response to mycobacteria residing within the macrophage phagolysosome.17 IL-12Rβ1 deficiency has an incomplete penetrance and age of manifestation, severity and outcome are highly variable [3, 5]. In the here presented case, the patient had not suffered any unusual, recurrent or severe infections in the past.
A strength of this case report is that, retrospectively, the established diagnosis can be clearly discriminated from sarcoidosis. We are confident that the patient did neither acquire the infection under the immunosuppressive treatment she initially received, nor that sarcoidosis was triggered by the infection. This case report is limited by its short follow-up period. At the time of writing, the patient is still receiving treatment and thus we cannot provide information on long-term outcome.
In this report we present a rare case of a disseminated infection with M. genavense owing to an IL-12Rβ1-associated immunodeficiency in an otherwise healthy 31-year-old female who was initially misdiagnosed with sarcoidosis. Likewise, long-term corticosteroid treatment may have resulted in a further impairment of immune control of mycobacterial infection and uncontrolled bacterial amplification. Sarcoid-like reaction does not equal sarcoidosis, and sarcoidosis remains a diagnosis of exclusion [1]. However, diagnosing latent or subclinical infections may be challenging when pathogens are hardly detectable or not expected in an apparently immunocompetent patient. Patients with MSMD may be asymptomatic until adulthood, and MSMD or alternative genetic defects of mycobacterial immune control should be suspected in patients with disseminated mycobacterial infection without pharmacological immunosuppression or acquired immunodeficiency.
Availability of data and materials
Not applicable.
Abbreviations
- ACE:
-
Angiotensin converting enzyme
- 18FDG-PET/CT:
-
18FDG-positron emission tomography/computed tomography
- HIV:
-
Human immunodeficiency virus
- IFN-γ:
-
Interferon gamma
- IL-12 :
-
Interleukin 12
- IL-12Rβ1:
-
Interleukin 12 receptor subunit beta 1
- iNOS:
-
Inducible nitric oxide synthase
- MSMD:
-
Mendelian susceptibility to mycobacterial diseases
- PCR:
-
Polymerase chain reaction
- sIL2R:
-
Soluble interleukin 2 receptor
- TNF :
-
Tumor necrosis factor
References
Drent M, Crouser ED, Grunewald J. Challenges of sarcoidosis and its management. N Engl J Med. 2021;385(11):1018–32. https://doi.org/10.1056/NEJMRA2101555.
Crouser ED, Maier LA, Baughman RP, et al. Diagnosis and detection of sarcoidosis. An Official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med. 2020;201(8):E26–51. https://doi.org/10.1164/RCCM.202002-0251ST.
Bustamante J, Boisson-Dupuis S, Abel L, Casanova JL. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-γ immunity. Semin Immunol. 2014;26(6):454–70. https://doi.org/10.1016/J.SMIM.2014.09.008.
Noma K, Mizoguchi Y, Tsumura M, Okada S. Mendelian susceptibility to mycobacterial diseases: state of the art. Clin Microbiol Infect. 2022. https://doi.org/10.1016/J.CMI.2022.03.004.
De Beaucoudrey L, Samarina A, Bustamante J, et al. Revisiting human IL-12Rβ1 deficiency: a survey of 141 patients from 30 countries. Medicine (Baltimore). 2010;89(6):381–402. https://doi.org/10.1097/MD.0B013E3181FDD832.
Böttger E, Hirschel B, Coyle M. Mycobacterium genavense sp. Nov. Int J Syst Bacteriol. 1993;43:841–3. https://doi.org/10.1099/00207713-43-4-841.
Mahmood M, Ajmal S, Abu Saleh OM, Bryson A, Marcelin JR, Wilson JW. Mycobacterium genavense infections in non-HIV immunocompromised hosts: a systematic review. Infect Dis (London, England). 2018;50(5):329–39. https://doi.org/10.1080/23744235.2017.1404630.
van de Vosse E, de Paus RA, van Dissel JT, Ottenhoff THM. Molecular complementation of IL-12Rbeta1 deficiency reveals functional differences between IL-12Rbeta1 alleles including partial IL-12Rbeta1 deficiency. Hum Mol Genet. 2005;14(24):3847–55. https://doi.org/10.1093/HMG/DDI409.
Karadaǧ A, Usta E, Bilgin K, Güney AK, Eroǧlu C, Günaydin M. Comparison of culture, real-time DNA amplification assay and Ehrlich-Ziehl-Neelsen for detection of Mycobacterium tuberculosis. Balkan Med J. 2013;30(1):13. https://doi.org/10.5152/BALKANMEDJ.2012.061.
Esteve-Solé A, Sologuren I, Martínez-Saavedra MT, et al. Laboratory evaluation of the IFN-γ circuit for the molecular diagnosis of Mendelian susceptibility to mycobacterial disease. Crit Rev Clin Lab Sci. 2018;55(3):184. https://doi.org/10.1080/10408363.2018.1444580.
Dumonceau JM, Fonteyne PA, Realini L, Van Gossum A, Van Vooren JP, Portaels F. Species-specific Mycobacterium genavense DNA in intestinal tissues of individuals not infected with human immunodeficiency virus. J Clin Microbiol. 1995;33(9):2514–5. https://doi.org/10.1128/jcm.33.9.2514-2515.1995.
Kiehn TE, Hoefer H, Bottger EC, et al. Mycobacterium genavense infections in pet animals. J Clin Microbiol. 1996;34(7):1840–2. https://doi.org/10.1128/jcm.34.7.1840-1842.1996.
Hillebrand-Haverkort ME, Kolk AHJ, Kox LFF, Ten Velden JJAM, Ten Veen JH. Generalized Mycobacterium genavense infection in HIV-infected patients: detection of the mycobacterium in hospital tap water. Scand J Infect Dis. 1999;31(1):63–8. https://doi.org/10.1080/00365549950161907.
Cornelis G, Reynders M, Deprez J, Vankeerberghen A, Orlent H. Disseminated Mycobacterium genavense infection in an immunocompetent adult: a case report. Clin Microbiol Infect. 2018;24(12):1355–6. https://doi.org/10.1016/j.cmi.2018.07.013.
Santos M, Gil-Brusola A, Escandell A, Blanes M, Gobernado M. Mycobacterium genavense infections in a tertiary hospital and reviewed cases in non-HIV patients. Patholog Res Int. 2014;204:1–8. https://doi.org/10.1155/2014/371370.
Brownell I, Ramiŕez-Valle F, Sanchez M, Prystowsky S. Evidence for Mycobacteria in Sarcoidosis. Am J Respir Cell Mol Biol. 2011;45(5):899. https://doi.org/10.1165/RCMB.2010-0433TR.
Weiss G, Schaible UE. Macrophage defense mechanisms against intracellular bacteria. Immunol Rev. 2015;264(1):182–203. https://doi.org/10.1111/IMR.12266.
Acknowledgements
We would like to thank the patient for giving us permission to publish her case and the hospital personnel who was involved in the diagnostic workup and treatment.
Funding
This article received no external funding.
Author information
Authors and Affiliations
Contributions
IT and GW conceptualized the case report. SD, SL and JF wrote the first draft of the manuscript. CG, AP, MS, JZ, RBW, GW and IT critically read the manuscript and provided important input. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Consent for publication
Written informed consent was obtained from the patient for the publication of this report and any accompanying images. Ethics approval was provided by the Ethics Committee of Innsbruck Medical University (EK Nr:1198/2022).
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Denicolò, S., Laydevant, S., Fink, J. et al. Sarcoid-like lesions obfuscating the diagnosis of disseminated Mycobacterium genavense infection in a patient with IL-12Rβ1-associated immunodeficiency. BMC Infect Dis 22, 770 (2022). https://doi.org/10.1186/s12879-022-07644-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-022-07644-4