
Abstract
Introduction
COVID-19 vaccines were rapidly authorised, thus requiring intense post-marketing re-evaluation of their benefit-risk profile. A multi-national European collaboration was established with the aim to prospectively monitor safety of the COVID-19 vaccines through web-based survey of vaccinees.
Methods
A prospective cohort event monitoring study was conducted with primary consented data collection in seven European countries. Through the web applications, participants received and completed baseline and up to six follow-up questionnaires on self-reported adverse reactions for at least 6 months following the first dose of COVID-19 vaccine (Netherlands, France, Belgium, UK, Italy) and baseline and up to ten follow-up questionnaires for one year in Germany and Croatia. Rates of adverse reactions have been described by type (solicited, non-solicited; serious/non-serious; and adverse events of special interest) and stratified by vaccine brand. We calculated the frequency of adverse reaction after dose 1 and prior to dose 2 among all vaccinees who completed at least one follow-up questionnaire.
Results
Overall, 117,791 participants were included and completed the first questionnaire in addition to the baseline: 88,196 (74.9%) from Germany, 27,588 (23.4%) from Netherlands, 984 (0.8%) from France, 570 (0.5%) from Italy, 326 (0.3%) from Croatia, 89 (0.1%) from the UK and 38 (0.03%) from Belgium. There were 89,377 (75.9%) respondents who had received AstraZeneca vaccines, 14,658 (12.4%) BioNTech/Pfizer, 11,266 (9.6%) Moderna and 2490 (2.1%) Janssen vaccines as a first dose. Median age category was 40–49 years for all vaccines except for Pfizer where median age was 70–79 years. Most vaccinees were female with a female-to-male ratio of 1.34, 1.96 and 2.50 for AstraZeneca, Moderna and Janssen, respectively. BioNtech/Pfizer had slightly more men with a ratio of 0.82. Fatigue and headache were the most commonly reported solicited systemic adverse reactions and injection-site pain was the most common solicited local reaction. The rates of adverse events of special interest (AESIs) were 0.1–0.2% across all vaccine brands.
Conclusion
This large-scale prospective study of COVID-19 vaccine recipients showed, for all the studied vaccines, a high frequency of systemic reactions, related to the immunogenic response, and local reactions at the injection site, while serious reactions or AESIs were uncommon, consistent with those reported on product labels. This study demonstrated the feasibility of setting up and conducting cohort event monitoring across multiple European countries to collect safety data on novel vaccines that are rolled out at scale in populations which may not have been included in pivotal trials.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The large scale roll-out of the novel COVID-19 vaccines called for the introduction of a monitoring system in multiple countries for rapid data collection. |
This study showed the feasibility of introducing cohort event monitoring in multiple countries and pooling of aggregated results allowed for near real-time monitoring of initial patient reported reactions. |
1 Introduction
The COVID-19 pandemic led to the accelerated marketing of newly developed vaccines against SARS-COV-2 with unprecedented speed, on the basis of conditional approval. For the COVID-19 vaccines, rapid speed, large Phase 2–3 clinical trials were organised. For the BioNTech/Pfizer vaccine, a multinational, placebo-controlled, observer-blinded, efficacy trial included 43,448 subjects: 21,720 with the BioNTech/Pfizer vaccine. The primary end points of this trial were solicited, specific local or systemic adverse events (AEs) and use of antipyretic or pain medication within 7 days after the receipt of each dose of vaccine or placebo. Unsolicited serious AEs were followed through 6 months after the second dose [1]. For the AstraZeneca vaccine, a multi-national randomised controlled trial enrolled 23,848 participants, 11,636 of whom were included in the interim primary efficacy analysis. The median follow-up period for AEs was 3.4 months after the first dose and 2.0 months after the second dose [2]. A comparative systematic review and meta-analysis of reactogenicity, immunogenicity and efficacy of COVID-19 vaccines, which included 32 studies, demonstrated a 1.7-fold increased risk of any AE in the vaccinated group compared to control groups in the trials. The majority of reported AEs in those studies were mild, and were local and systemic [3]. The vaccine manufacturers that received initial conditional approvals by the European Medicines Agency (EMA) were BioNTech/Pfizer [4].
The lack of extended follow-up and of inclusion of fragile persons (e.g. pregnant and lactating women, immunocompromised patients, patients with history of allergy and SARS-COV-2 infection, etc.) in the pivotal clinical trials as well as the scale of the vaccination campaigns made it imperative to closely monitor COVID-19 vaccine safety during the roll-out of the vaccine [5]. In the post-authorisation phase, safety monitoring is typically conducted through spontaneous reporting systems, which collect data on suspected adverse drug reactions (ADRs) that are spontaneously reported by healthcare professionals and citizens. In Europe, reports of ADRs are collected nationally and sent to the EudraVigilance system of the EMA. Analysis of the data, alone or in combination to other pharmacovigilance data-sources, can lead to the detection of signals [9], but not allow to clearly estimate prevalence of ADRs, since spontaneous reporting, by nature, is not exhaustive.
Cohort event monitoring (CEM) is considered active surveillance and allows for in-depth information on the time course of reported ADRs, with a clear denominator allowing risk quantification. It is well suited to capture reactogenic events, including those that are not medically attended. The value of CEM as complement to the existing spontaneous reporting systems was shown during the H1N1 pandemic in different European countries. For instance; a CEM study in the Netherlands gave insight into the incidence of adverse reactions in a large group of vaccinated persons over a period of months and it was possible to follow the time course, information about time to onset, duration of reactions and action taken when experiencing an adverse reaction. These data were under-represented in published trials [6].
Cohort event monitoring studies of COVID-19 vaccines have been rolled out in both the USA and the UK. The V-Safe study in the USA uses a smartphone-based vaccine monitoring system where vaccinated persons answer web-based questionnaires for post-marketing vaccine safety surveillance [7]. In the UK, data from vaccinated persons have been collected through the COVID Symptom Study app. The app enables self-reported information related to SARS-CoV-2 infection to be captured. Vaccinated persons were asked to register data on their vaccination and the occurrence of adverse reactions [8]. In the COVID-19 Citizen Science Study, an online cohort study, participants completed surveys on health and COVID-19–related events [9].
The Early COVID-19 Vaccine Monitor (ECVM) CEM project aimed to prospectively collect adverse reaction data in near real time from a large cohort of COVID-19 vaccine recipients in different European countries. A multinational CEM study provides the opportunity to gather safety data rapidly and to monitor novel vaccines when they roll out with a well-defined denominator.
2 Methods
2.1 Design
Data on occurrence of self-reported, suspected ADRs following the administration of marketed COVID-19 vaccines were collected through a multi-site prospective CEM. Vaccinees were invited to participate and could register to the web-based app up to 48 hours after receiving the first COVID-19 vaccine dose. Participants were required to provide informed consent during registration and complete a baseline questionnaire to collect information on gender, age, comorbidities, concomitant drugs, and COVID-19 vaccination. Participants in Germany were prompted to report comorbidities after one year, rather than at baseline. Follow-up questionnaires were sent after the baseline questionnaire.
2.2 Sites
Participants from seven European countries were included in this study: Belgium, Croatia, France, Germany, Italy, the Netherlands and the UK. Organisations contributing to the CEM project were either National Competent Authorities (NCAs), were working in close collaboration with an NCA, or received support from an NCA. Table 1 summarises the contributing organisations by country.
2.3 Data Collection
Three countries had started preparations for national CEM using their own data collection systems prior to the ECVM study start: Germany in December 2020, Netherlands and Croatia in February 2021. Other countries started using the Dutch system (see Table 1). Data collection systems were aligned, as much as possible, with countries through the use of one common protocol with a predefined set of questions. Agency for Medicinal Products and Medical Devices of Croatia (HALMED) used a web-based application called OPeN (Online Platform for Electronic reporting of adverse drug reactions), adapted for the purpose of CEM in Croatia by providing access to all vaccinees and including scheduled questionnaires. In Germany, the Paul-Ehrlich-Institute used the smartphone application SafeVac 2.0, initially designed to monitor the safety of annual influenza vaccines and adapted to record safety of the COVID-19 vaccines. The other five participating countries used the Lareb Intensive Monitoring (LIM) web app, developed by Lareb in the Netherlands for CEM. Participants could register themselves on a website designed specifically for this study and each organisation had a country-specific website and questionnaires. Approvals from ethics committees were obtained in all countries and questionnaires were translated to local language and were tested.
2.4 Follow-up Questionnaires
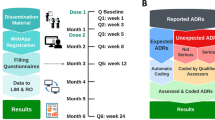
Participants received up to six follow-up questionnaires with questions related to possible adverse reactions experienced after receiving the first and second dose of the COVID-19 vaccine over a period of 6 months (all countries using LIM) or up to ten questionnaires over 12 months (Croatia, Germany) after first vaccine dose administration. The questionnaires were scheduled to ensure that short-term data on possible adverse reactions could be collected after the first and second dose of the vaccine in each of the countries and that additional follow-up information on medium- to long-term safety was collected around 3 and 6 months after the first vaccination date (Fig. 1). This vaccination date was reported by the participant and questionnaire scheduling was anchored for this date.

Questionnaire scheduling schemes for the participating countries over time (days). b baseline, LIM data collection tool used by the Netherlands, Italy, Belgium, France and the UK with three different schedules per country, OPeN data collection tool used by Croatia, q questionnaire, SafeVac 2.0 data collection tool used by Germany with different schedule per vaccine brand
It was expected that most ADRs occurred within 72 h after vaccination and the most well-known ADRs recovered within five days of vaccination. Therefore, the first questionnaires were sent in the first and second week after vaccination as shown in Fig. 1. To get the most accurate information on ADRs after the second dose, questionnaires were sent near to the expected date of vaccination dose 2, based on a minimum interval of four weeks between first and second dose. The schedules used for follow-up questionnaires were adapted as far as possible to vaccine brand and country-specific scheduled 1–2 dose administrations and as such could differ per country (Fig. 1).
2.5 Type of Adverse Reactions
Outcomes of interest were local and systemic reactogenic adverse reactions, adverse events of special interest (AESIs) [10] and other serious adverse reactions, which were judged by the vaccinee to be related to vaccination. Based on AEs most frequently reported in pivotal trials [1, 2, 11, 12], the following events were solicited: fatigue, headache, malaise, chills, myalgia, fever, arthralgia, nausea and injection-site reactions. Germany included two additional solicited adverse reactions: diarrhoea and dizziness. Participants were systematically asked whether they experienced these reactions; however, they were also able to report any other suspected adverse reactions. All reactions were coded using the MedDRA coding system (version 24.0) [13].
All data collection tools were built to include coding of the data, both manually and automatically. The solicited ADRs were coded with their corresponding Preferred Terms (PT), except fever and injection-site reaction, which had more extensive options for coding. These solicited ADRs were coded automatically in the SafeVac 2.0 app and the LIM web app as described in Fig. 2. Coding of unsolicited reactions was guided by MedDRA points to consider [14] and internal guidance documents to maintain coding consistency. Trained assessors in the participating countries coded the reported ADRs into English Lower-Level Terms (LLT).

Flowchart of adverse drug reaction (ADR) assessment in systems with automatic coding
In this way, a library was created in which participant-reported text was linked to the MedDRA code chosen by an assessor. From this library, MedDRA code suggestions were assigned automatically to future reported unsolicited reactions in the same language and country. This process of auto-coding helped improve data quality and minimise time and resources needed for coding.
According to Council for International Organizations of Medical Sciences (CIOMS) criteria [15], reported reactions were classified as serious if: fatal, life-threatening, causing/prolonging hospitalisation, resulting in persistent or significant disability or incapacity, requiring intervention to prevent permanent damage, or causing congenital anomalies. The assessors also manually evaluated whether the reported ADR, solicited or unsolicited, met the CIOMS seriousness criteria and, when necessary, requested additional information from the vaccine. Adverse events of special interest were defined based on the list of AESI (see Electronic Supplementary Material 2) that was established by the ACCESS project during the initiation of the study [16]. Adverse reactions reported in questionnaires between the date of the first dose and the date of the second dose were defined as dose 1 adverse reactions.
2.6 Data Sharing
Germany and Croatia shared aggregated data from their own applications with Lareb by local completion of pre-defined shell tables containing: age, gender, country, comorbidity, reported adverse reactions, vaccine brand and whether data were reported between the first and second dose or after the second dose. Data reported and assessed up to October 2021 are included in this paper. Release of data in Germany was organised by manufacturer. For the current paper it comprises data on AstraZeneca and Moderna vaccine only, as data on BioNTech/Pfizer were incomplete at time of writing. Data collected in the LIM app by the various countries were submitted to the Lareb server and analysed by Lareb. Monthly updates based on aggregated data from all study sites were pushed to an interactive POWERBI dashboard for use by consortium members and the EMA. In addition, all reactions reported through the data collection tools in Belgium, Croatia, Germany, Italy and the Netherlands were submitted to the EudraVigilance (EV) database. Data processing is described in Electronic Supplemental Material 1.
2.7 Data Analysis
The data available for this report were aggregated datasets with predefined stratification. We used descriptive statistics to describe baseline characteristics of participating vaccinees. Recruited participants were defined as persons registering for the study. Responding participants were defined as persons registering for the study and providing baseline information and at least one questionnaire related to dose 1. Percentage of adverse reactions was calculated using the number of subjects reporting an adverse reaction in questionnaires related to dose 1, divided by the number of subjects who completed any of the questionnaires related to dose 1.
3 Results
A total of 216,260 subjects registered for the CEM and 117,791 (54.5%) of the recruited participants completed at least one questionnaire after dose 1; of those, 29,270 (24.8%) persons were participating through the LIM app, 326 (0.3%) the OPeN app (Croatia) and 88,197 (74.9%) in SafeVac 2.0 (Germany).
Of the 117,791 participants, 89,377 (75.9%) had received a first dose of AstraZeneca vaccine, 14,658 (12.4%) a first dose of BioNTech/Pfizer vaccine, 11,266 (9.6%) had a first dose of the Moderna vaccine and 2490 (2.1%) participants received a first dose of the Janssen vaccine (Table 2). Median age category was between 40 and 49 years for respondents having AstraZeneca, Moderna or Janssen Covid-19 vaccine, and 70–79 years for BioNTech/Pfizer median age category (Table 2). The majority of vaccinees with AstraZeneca vaccine dose 1 were female (57.2%) and for Janssen this was 71%. For vaccinees with Pfizer vaccine 45.1% were female and for Moderna 68.2% of gender was unknown. This was due to technical difficulties in data extraction from Germany.
3.1 Solicited Adverse Reactions
The most commonly reported solicited systemic reaction was fatigue, which was closely followed by headache and malaise (Table 3). Fatigue was reported by 48,212 (53.9%) participants who received the AstraZeneca vaccine and 1261 (50.6%) participants who received Janssen. Fewer participants reported this adverse reaction after their first dose of BioNTech/Pfizer and Moderna vaccines, in total 2519 (17.2%) and 3971 (35.2%) participants, respectively. Headache was reported almost as often as fatigue in those receiving AstraZeneca and Janssen, 53.3% and 49.6%, respectively. This was lower in the BioNtech/Pfizer (11.9%) and Moderna (29.8%) groups. It was found that reports of any systemic solicited reactions were more frequent in participants receiving the AstraZeneca or Janssen vaccine compared with participants receiving other vaccine brands. Participants who received the BioNtech/Pfizer vaccine were less likely to report any systemic solicited adverse reactions when compared with other vaccine brands. The most commonly reported local reaction across all vaccine brands was coded as ‘injection-site pain’ with participants receiving AstraZeneca and Moderna reporting this reaction most frequently, 47,187 (52.8%) and 5755 (51.1%) participants, respectively. For participants receiving Moderna, this was overall the most commonly reported solicited adverse reaction. Participants who received BioNTech/Pfizer reported ‘injection-site pain’ at a higher rate (24.1%) than any of the systemic solicited adverse reactions.
3.2 Non-solicited Reactions
Dizziness was the most frequently reported unsolicited adverse reaction across all vaccine brands A total of 17,051 (19.1%) participants receiving AstraZeneca reported this reaction, followed by 1001 (8.9%) participants receiving Moderna. Only 158 (1.1%) and 56 (2.3%) participants receiving BioNTech/Pfizer and Janssen, respectively, reported dizziness. Diarrhoea was the second most commonly reported unsolicited reaction, with AstraZeneca and Moderna recipients showing the most frequent reports of 4.1% and 3.6%, respectively. These two events were included as solicited adverse reactions in Germany, which may have increased the frequency of reporting these reactions. Other than these two adverse reactions, the frequency and type of other non-solicited adverse reactions was lower and varied highly across vaccine brands, with more than 800 different MedDRA Preferred Terms being coded. Table 4 describes the 10 most frequently reported unsolicited adverse reactions.
3.3 AESI and Serious Adverse Reactions
Of the 2490 participants who received Janssen, 5 (0.2%, 95% CI 0–0.4%) reported an adverse reaction that was classified as AESI, versus 7 (0.1%, 95% CI 0–0.1%) for Moderna, 20 (0.1%, 95% CI 0–0.2%) for BioNTech/Pfizer and 95 (0.11%, 9% CI 0.10–0.12%) for AstraZeneca. The most commonly reported AESI was respiratory distress, which was observed in 44, 3, and only 1 in AstraZeneca, Moderna and BioNtech/Pfizer vaccinated respondents, respectively (Table 5). Of the 89,337 participants who received AstraZeneca, 628 (0.7%, 95% CI 0.6–0.8%) reported at least one serious adverse reaction, as compared to 45 (0.4%, 95% CI 0.3–0.5%) for Moderna, 4 (0.2%, 95% CI 0–0.3%) for Janssen and 16 (0.1%, 95% CI 0.1–0.2%) for BioNTech/Pfizer participants.
4 Discussion
Setting up and conducting this CEM study in seven countries was a challenging task, but this study shows the feasibility of such an undertaking. Data were made available in real-time on dashboards, allowing for regular updates on adverse reaction rates as the vaccines were being rolled out across Europe. The results presented should be interpreted in the light of vaccination recommendations at the time of data capture. Due to the aggregated nature of these initial datasets used, additional stratification and analyses were not possible; however, this will change as more data become available and analyses can be performed on record-based rather than aggregated data. Nevertheless, the following insights gained on solicited, unsolicited and serious adverse reactions along with monitoring of AESI provided important safety data in a rapid context.
4.1 Solicited Adverse Reactions
The solicited reactions are expected and labelled, these ADRs are directly related to the immune response that is boosted by the vaccine or the injection site. Across all vaccine brands, fatigue was the most commonly reported solicited systemic adverse reaction by study participants: 48,212 (53.9%), 2519 (17.2%), 1261 (50.6%) and 3971 (35.3%) for AstraZeneca, BioNtech/Pfizer, Janssen and Moderna, respectively. Headache was the second most frequent ADR for AstraZeneca with 47,640 (53.3%), Janssen with 1236 (49.6%) and Moderna with 3353 (29.8%) participants reporting this reaction. However, for BioNtech/Pfizer recipients, myalgia was the second most commonly reported solicited adverse reaction. These findings are in accordance with the Summary of Product Characteristics (SmPCs) for these vaccines [4, 17,18,19]. The most common AEs described for BioNtech/Pfizer [4] include pain and swelling at the injection site, tiredness, headache, muscle and joint pain, chills, fever and diarrhoea. They affected more than 10% of vaccinees and were usually mild or moderate and improved within a few days after vaccination. This is comparable to the rates reported in this study for fatigue (tiredness), headache and myalgia (muscle ache); however, arthralgia (joint pain) and injection-site swelling were reported less frequently, 581 (3.9%) and 727 (5.0%), respectively. Due to the aggregated nature of the datasets used, adjusting for age and gender was not possible, particularly for BioNtech/Pfizer where more than 50% of data were represented by participants aged > 70 years. For Moderna [17] adverse reactions labelled as common were pain and swelling at the injection site, tiredness, chills, fever, swollen or tender lymph nodes under the arm, headache, muscle and joint pain, nausea (feeling sick) and vomiting. For AstraZeneca [18] common adverse effects listed were tenderness, pain and bruising at the injection site, headache, tiredness, muscle pain, general feeling of being unwell, chills, fever, joint pain and nausea. For Janssen [19], the adverse reactions labelled as common were pain at the injection site, headache, tiredness, muscle pain and nausea. In our study, fatigue and headache were both reported in more than 50% of participants after the first dose of AstraZeneca vaccine and in approximately 50% of Moderna recipients. Fever was reported by more than 30% of participants who received AstraZeneca and Moderna—higher than those reported for the Pfizer and Janssen vaccines. For Pfizer and Janssen, the percentage of vaccinated persons who reported pyrexia was lower than in the SmPC, which may be due to age and co-morbidity (and immunogenic response) between respondents and trial participants.
4.2 Non-solicited Adverse Reactions
Dizziness, diarrhoea and vomiting were not in the predefined list of solicited reactions for most countries in our study, except for Germany where dizziness and diarrhoea were solicited adverse reactions. These were frequently reported systemic effects for all vaccine brands. Dizziness was reported most often by participants receiving the AstraZeneca vaccine (19.1%). This is a higher percentage than is currently presented in the SmPC; the frequency is labelled ‘Uncommon (may affect up to 1 in 100 people)’ [18]. However, dizziness was a solicited event in Germany. The AstraZeneca vaccine recipients (almost all from Germany) reported many common solicited adverse reactions: half of all respondents reported fatigue and headache while more than one-third reported malaise, chills, myalgia and pyrexia. Half of all respondents receiving the Janssen vaccine reported fatigue, headache and malaise and one-third experience chills, myalgia and pyrexia.
4.3 AESI and Serious Adverse Reactions
Adverse events of special interest were rare, which is consistent with the product labels. The SmPC of Moderna reports sleepiness as being a very common (> 1/10) reaction with no further mention of severe forms as reported in this study: 1 (0.009%) participant reported hypersomnia [17]. Facial swelling was one of the few rare AESIs reported in the SmPC of Moderna, but was not reported by any participants receiving this vaccine brand. The majority of AESIs were reported by participants receiving AstraZeneca, with most being for respiratory distress: a total of 40 (0.05%) participants reported this reaction. This is not in line with the SmPC for AstraZeneca, as there is no mention of pulmonary disorders reported post-vaccination [18]. Further analysis on age and comorbidities of these participants is necessary in order to understand whether or not this is related to underlying health issues. Many thrombotic events are reported by participants who received AstraZeneca. For example, 1 (0.001%) participant reported an acute myocardial infarct and 2 (0.002%) participants reported pulmonary embolism. The SmPC for AstraZeneca only mentions thrombotic events in combination with thrombocytopenia [18].
Serious reactions in this study did not give rise to a safety signal on their own but were used in routine signal detection procedures, next to spontaneous reports.
The rate of serious reactions was uncommon in all recipients of all vaccine brands between first and second doses. The crude rate was highest for AstraZeneca recipients with 0.7%, 95% CI 0.6%–0.8%, followed by 0.4%, (95% CI 0.3–0.5%) for Moderna, 0.2%, (95% CI 0–0.3%) for Janssen and 0.11%, (95% CI: 0.10–0.2%) for BioNTech/Pfizer participants. Whether these differences can be explained by the age/gender/co-morbidity distribution of the population receiving this vaccine, media attention around vaccines or higher risks around these vaccines cannot be answered based on the aggregated data. Future analyses will require standardisation and/or adjustments for baseline characteristics to allow for comparison of rates between vaccine brands.
4.4 Strengths and Limitations
The strength of this study is the large cohort size, which spans across multiple European countries and collects detailed and longitudinal data on safety of the novel COVID-19 vaccines. The use of patient-reported outcomes enables us to capture data on adverse reactions that would otherwise not be captured in medical records because most vaccinated persons who have short-lasting, non-serious adverse reactions, will not attend their physician. Cohort event monitoring studies have earned their place among the methods of gathering rapid post-marketing data on safety, especially in the first phase of roll-out when large administrative data sources that can be used retrospectively, are not yet available, or in countries where these are not available at all [6, 20,21,22,23].
This study has several limitations, some of these being temporary, while others are inherent to the setting of the study. First, the study is ongoing and we report follow-up data up until the time this paper was drafted. Second, participants who experienced an adverse reaction may be more inclined to fill in follow-up questionnaires. However, the limitation of the registration within 48 hours after receiving the vaccination should partially limit this issue. The 48 hours were necessary since most participants would only be invited to participate at vaccination sites. Participants who experienced an adverse reaction could be more likely to continue their participation longer than those who experienced no adverse reactions. However, a previous study for the Dutch data reported the results of a sensitivity analysis, which showed that this bias was limited [24].
Reporting may depend on age, and crude comparisons between brands are confounded by age and other variables. Respondents with BioNTech/Pfizer vaccine were much older than respondents with other vaccines, which may have led to the lower rate of adverse reactions since the immune response (and therefore reactogenicity) may be lower with old age [25]. Confounding by gender, age and co-morbidity was not accounted for in this first paper. The start date of the studies had an impact on the absolute number of participants and inclusions because of the rapid roll-out of the vaccination campaigns. The willingness for the general public to contribute to vaccine safety was probably higher at the start of vaccine roll-out, which may be a disadvantage to the sites that started the study much later than initially planned, despite efforts to promote the studies and disseminate study material to increase inclusions. The willingness to continue participation may have decreased over time, with a considerable loss to follow-up. In turn this would lead to bias in the data collected in later rounds. Analysing this loss to follow-up with an additional comparison between countries will be necessary to understand the impact. We decided that the use of the persons with at least one follow-up questionnaire as denominator may lead to an overestimate of the rate of adverse reactions, which we did to be conservative.
All data collection tools were internet- or smartphone-based applications. Only those participants with an internet connection and/or a smartphone were able to register for the study. While countries with multiple official languages had the opportunity to have multi-language data collection tools, it was not possible to offer other language options for smaller groups such as migrants. Participants had to fill in their questionnaires by themselves or find help independently to do so. Despite these limitations, the elderly population does seem to be represented across multiple applications, countries and vaccine brands.
The limitation of the aggregated data and the availability of updated data leads to data which may not be representative of the vaccinated population. Additionally, comparisons, whether between vaccine brands or between countries, are only possible when confounding factors such as age, gender and comorbidities, can be considered. With the addition of the data for future analyses, data on other subpopulations may become available.
The roll-out of the novel COVID-19 vaccines was rapid and with every approval of a new vaccine brand, European countries were able to add a new vaccine to their vaccination strategies. Data collection had to be adapted along with all these changes, including adding additional vaccine brands with different dose intervals. Unfortunately, the roll-out of the LIM data collection tool was much later in some countries than the roll-out of the vaccines, leading to certain populations that were vaccinated early on in the process, not being included. It points to the fact that for future monitoring, readiness of data collection infrastructures and approvals is important to implement CEM. Those countries with readiness of data collection infrastructures in place, funded by national governments, were ready to start enrolment at the beginning of the vaccination campaign.
In addition, the variation in interpretation of General Data Protection Regulation (GDPR) in Europe lead to a delay in medical and ethical committee approval, which in turn severely delayed the start of the study for many countries. The ECVM study has given insight into this variation. Medical and ethical approval should be initiated as early as possible.
Vaccine safety was a concern for the general public and extensive media attention was given when new information on vaccine safety became available. This media attention may have impacted on the reporting of adverse reactions, as observed in the spike of reports to spontaneous reporting systems [26]. In April 2021, EMA confirmed a possible link between AstraZeneca and the rare combination of blood clots combined with low platelet counts [27], which lead to a (temporary) halt in vaccinating specific sub-populations with this vaccine [28]. This also meant that the targeted population for specific vaccine brands was adjusted during data collection.
For this study, data on adverse reactions occurring between the first and second vaccination were included. The follow-up period following dose 1 is comparable across all countries and all data collection systems. Due to varying schedules of the questionnaires, combined with variations in the dose interval of the vaccine brands, and the changes made in these intervals in the national vaccination strategies, analysis of dose 2 data without taking these points into account is not reliable and was therefore not included. Future analysis will provide insight into the safety profile after the second dose of vaccination or during longer term follow-up.
4.5 Future Studies
The rapid introduction of the novel COVID-19 vaccines along with the many unknowns such as varying national vaccination strategies, unknown dose intervals, and target populations were challenging for the study initiation. Avoiding these unknowns in future may be impossible; however, the ECVM study gives insight into these obstacles for future pandemic preparedness.
The ECVM study has provided real-time monitoring in the early phase of COVID-19 vaccine distribution and has created valuable data for further analysis. It allowed access to near-real time data for regulators through a dashboard. Moreover, reactions were also reported to the EudraVigilance data source, which can be queried by the public. The longitudinal character of the data collection will make it possible to explore time patterns of reactions and the occurrence of reactions with a longer time-to-onset. The role of age, sex and comorbidities and concomitant use of medication in the occurrence of adverse effects needs to be further explored. This should be done in light of the different study populations included for the various countries, with different vaccination strategies.
5 Conclusion
This study, that collates person-reported data from more than 100,000 COVID-19 vaccine recipients, showed that it is possible to harmonise CEM across multiple countries to collect safety data on novel vaccines that are rolled out at scale in populations that may not have been included in pivotal trials. The study confirmed the safety information in the product labels: reactogenic reactions, related to immunogenic response, and local injection-site reactions were very common across all vaccines, whereas serious reactions or AESIs were rare. Dizziness, diarrhoea and vomiting were the most frequent common non-solicited systemic reactions. Post-marketing monitoring of adverse reactions of the newly developed COVID-19 vaccines is not only necessary to understand the safety of these vaccines, but also gives insight into the more common and expected adverse reactions. Data on reported reactions will help inform vaccine recipients of the reactions they could expect and how these may differ by vaccine brand, and sex and age of the recipient.
References
Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N Engl J Med. 2020;383(27):2603–15. https://doi.org/10.1056/NEJMoa2034577.
Voysey M, Clemens SAC, Madhi SA, Weckx LY, Folegatti PM, Aley PK, et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet. 2021;397(10269):99–111. https://doi.org/10.1016/S0140-6736(20)32661-1.
McDonald I, Murray SM, Reynolds CJ, Altmann DM, Boyton RJ. Comparative systematic review and meta-analysis of reactogenicity, immunogenicity and efficacy of vaccines against SARS-CoV-2. NPJ Vaccines. 2021;6(1):74. https://doi.org/10.1038/s41541-021-00336-1.
European Medicines Agency (EMA). SmPC Comirnaty. 2022. https://www.ema.europa.eu/en/documents/product-information/comirnaty-epar-product-information_en.pdf Accessed 14 July 2022.
Dhanda S, Osborne V, Lynn E, Shakir S. Postmarketing studies: can they provide a safety net for COVID-19 vaccines in the UK? BMJ Evid Based Med. 2022;27(1):1–6. https://doi.org/10.1136/bmjebm-2020-111507.
Harmark L, van Hunsel F, Hak E, van Grootheest K. Monitoring the safety of influenza A (H1N1) vaccine using web-based intensive monitoring. Vaccine. 2011;29(10):1941–7. https://doi.org/10.1016/j.vaccine.2010.12.123.
Chapin-Bardales J, Gee J, Myers T. Reactogenicity Following Receipt of mRNA-Based COVID-19 Vaccines. JAMA. 2021;325(21):2201–2. https://doi.org/10.1001/jama.2021.5374.
Menni C, Klaser K, May A, Polidori L, Capdevila J, Louca P, et al. Vaccine side-effects and SARS-CoV-2 infection after vaccination in users of the COVID Symptom Study app in the UK: a prospective observational study. Lancet Infect Dis. 2021;21(7):939–49. https://doi.org/10.1016/S1473-3099(21)00224-3.
Beatty AL, Peyser ND, Butcher XE, Cocohoba JM, Lin F, Olgin JE, et al. Analysis of COVID-19 Vaccine Type and Adverse Effects Following Vaccination. JAMA Netw Open. 2021;4(12):e2140364. https://doi.org/10.1001/jamanetworkopen.2021.40364.
Brighton Collaboration. COVID-19 AESI list. 2020. https://brightoncollaboration.us/wp-content/uploads/2021/01/COVID-19-updated-AESI-list.pdf. Accessed 11 Oct 2021.
Baden LR, El Sahly HM, Essink B, Kotloff K, Frey S, Novak R, et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N Engl J Med. 2021;384(5):403–16. https://doi.org/10.1056/NEJMoa2035389.
Sadoff J, Gray G, Vandebosch A, Cárdenas V, Shukarev G, Grinsztejn B, et al. Final analysis of efficacy and safety of single-dose Ad26COV2.S. N Engl J Med. 2022;386(9):847–60. https://doi.org/10.1056/NEJMoa2117608.
The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Welcome to MedDRA. 2021. https://www.meddra.org/. Accessed 14 July 2022.
The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. MedDRA® POINTS TO CONSIDER COMPANION DOCUMENT ICH-Endorsed Guide for MedDRA Users. 2022. https://admin.meddra.org/sites/default/files/guidance/file/000515_companionptc_r2_0_Oct2020.pdf Accessed 28 Jan 2022.
CIOMS Working Group VIII. Practical Aspects of Signal Detection in Pharmacovigilance: Report of CIOMS Working Group VIII. Geneva2010. Report No.: 9290360828.
Willame C, Dodd C, Carlos D, Roel E, Gini R, Bartolini C, et al. Background rates of 41 adverse events of special interest for COVID-19 vaccines in 10 European Healthcare Databases—an ACCESS Cohort Study. SSRN Electron J. 2022. https://doi.org/10.2139/ssrn.4036376.
European Medicines Agency (EMA). SmPC Spikevax. 2022. https://www.ema.europa.eu/en/documents/product-information/spikevax-previously-covid-19-vaccine-moderna-epar-product-information_en.pdf. Accessed 14 July 2022.
European Medicines Agency (EMA). SmPC Vaxzevria 2022. https://www.ema.europa.eu/en/documents/product-information/vaxzevria-previously-covid-19-vaccine-astrazeneca-epar-product-information_en.pdf Accessed 14 July 2022.
European Medicines Agency (EMA). SmPC Covid-19 vaccine Janssen (Jcovden). 2022. https://www.ema.europa.eu/en/documents/product-information/covid-19-vaccine-janssen-epar-product-information_en.pdf. Accessed 14 July 2022.
Harmark L. Web-based Intensive Monitoring; a patient based pharmacovigilance tool (Thesis). Rijksuniversiteit Groningen; 2012.
Zhou L, Harrison-Woolrych M, Coulter DM. Use of the New Zealand Intensive Medicines Monitoring Programme to study the levonorgestrel-releasing intrauterine device (Mirena). Pharmacoepidemiol Drug Saf. 2003;12(5):371–7. https://doi.org/10.1002/pds.875.
Mackenzie IS, MacDonald TM, Shakir S, Dryburgh M, Mantay BJ, McDonnell P, et al. Influenza H1N1 (swine flu) vaccination: a safety surveillance feasibility study using self-reporting of serious adverse events and pregnancy outcomes. Br J Clin Pharmacol. 2012;73(5):801–11. https://doi.org/10.1111/j.1365-2125.2011.04142.x.
van Balveren-Slingerland L, Kant A, Harmark L. Web-based intensive monitoring of adverse events following influenza vaccination in general practice. Vaccine. 2015;33(19):2283–8. https://doi.org/10.1016/j.vaccine.2015.03.014.
Kant A, Jansen J, van Balveren L, van Hunsel F. Description of frequencies of reported adverse events following immunization among four different COVID-19 vaccine brands. Drug Saf. 2022;45(4):319–31. https://doi.org/10.1007/s40264-022-01151-w.
Pinti M, Appay V, Campisi J, Frasca D, Fulop T, Sauce D, et al. Aging of the immune system: Focus on inflammation and vaccination. Eur J Immunol. 2016;46(10):2286–301. https://doi.org/10.1002/eji.201546178.
Ferner RE, Stevens RJ, Anton C, Aronson JK. Spontaneous reporting to regulatory authorities of suspected adverse drug reactions to COVID-19 vaccines over time: the effect of publicity. Drug Saf. 2022. https://doi.org/10.1007/s40264-021-01138-z.
European Medicines Agency (EMA). Vaxzevria: further advice on blood clots and low blood platelets. 2021. https://www.ema.europa.eu/en/news/vaxzevria-further-advice-blood-clots-low-blood-platelets. Accessed 14 July 2022.
Wise J. Covid-19: European countries suspend use of Oxford-AstraZeneca vaccine after reports of blood clots. BMJ. 2021;372:n699. https://doi.org/10.1136/bmj.n699.
Acknowledgements
The authors acknowledge: Leontine van Balveren, Loes Ruijs and Jasper Schmitz for their contribution to the Early COVID-19 Vaccine Monitor (ECVM) study conducted in the Netherlands; Stéphanie Lamarque, Estelle Guiard, Emmanuelle Bignon, Pauline Bosco-Levy, Patrick Blin et Cécile Droz-Perroteau for their contribution to the French part of the ECVM study, EVANESCO; Ugo Moretti, Chiara Bellitto, Francesco Ciccimarra, Laura Augusta Gonella, Giuliana Petrelli, Cristiano Chiamulera and Elena Arzenton (University of Verona, Department of Diagnostics and Public Health – Section of Pharmacology, Verona, Italy); Riccardo Lora, David Bellantuono and Alberto Sabaini (MedBrains); Alberto Firenze, Donatella Zodda, Fabrizia Guidotti, Maria Zappone and Bernardo Alagna (Struttura Commissariale per l’Emergenza COVID della città metropolitana di Messina/Messina Local Health Unit, Messina, Italy); Paola Maria Cutroneo and Claudia Minore (Sicilian Regional Pharmacovigilance Centre, University Hospital of Messina, Messina, Italy); Claudio Costantino and Francesco Vitale (University of Palermo, Palermo, Italy – Department of Health Promotion, Mother and Child Care, Internal Medicine and MedicalSpecialties "G. D'Alessandro", Hygiene section); Ilaria Morreale – Sicilian Regional Center of Pharmacovigilance, Azienda Ospedaliera Universitaria Policlinico P. Giaccone, Internal Medicine, Pharmacovigilance and Clinical Pharmacology Unit; Laura Marsala, Desirè Farinella and Silvana Bavetta (Azienda Ospedaliera di Rilievo Nazionale e di Alta Specializzazione (ARNAS) “Civico”, Palermo, Italy); Maria Pia Fantini, Chiara Reno, Emanuel Raschi and Elisabetta Poluzzi (Alma Mater Studiorum-University of Bologna, Italy – Department of Medical and Surgical Sciences); Ester Sapigni, Annamaria Potenza, Debora Podetti, Victoria Nikitina, Rita Ricciardelli, Nazanin Mogheiseh, Silvia Croce and Barbara Paltrinieri (Emilia-Romagna Pharmacovigilance Regional Centre, Bologna – Italy); Sofia Castellani, Elisa Sangiorgi, Margherita Selleri, Simona Lucchesi and Giuseppe Catucci (Ferrara Local Health Unit, Ferrara, Italy); Denis Savini, Chiara Sacripanti, Marco Faccioli, Maria Silvia Romio and Laura Rossi (Bologna Local Health Unit, Bologna, Italy); Simonetta Radici – Piacenza Local Health Unit, Piacenza, Italy; Giovanna Negri - Parma Local Health Unit, Parma, Italy; Lidia Fares – Reggio Emilia Local Health Unit, Reggio Emilia, Italy; Chiara Ajolfi – Modena Local Health Unit, Modena, Italy; Antonella Fadda, Antonella Chiarello – Imola Local Health Unit, Imola, Italy; Fabio Pieraccini, Barbara Gavioli, Simonetta Palazzi – Romagna Local Health Unit, Romagna, Italy; Marco Tuccori – Tuscany Region, Italy – Unit of Adverse Drug Reactions Monitoring, University Hospital of Pisa, Unit of Adverse Drug Reactions Monitoring, Pisa, Italy; Alfredo Vannacci, Roberto Bonaiuti, Claudia Ravaldi, Niccolò Lombardi, Giada Crescioli – PeaRL – Perinatal Research Laboratory, NEUROFARBA Department, University of Florence and CiaoLapo Foundation for Perinatal Health; Florence, Italy; Francesco Gori – Firenze Local Health Unit, Florence, Italy; Roberto Tessari (IRCCS Ospedale Sacro Cuore Don Calabria – Hospital Pharmacy, Negrar di Valpolicella, Italy); Emanuela Zandonà (University Hospital of Verona, Italy – Medical Coordination Unit); Maria Angiola Crivellaro – University Hospital of Padua, Padova, Italy – Occupational Health Unit and Allergology Unit, Department of Cardiac Thoracic Vascular and Public Health Sciences University of Padova; Mauro Cancian – University Hospital of Padua, Departmental Allergy Unit, Padua, Italy; Francesca Venturini – University Hospital of Padua, Pharmacy Department, Padua, Italy; Marina Ferri and Luca Leonardi (Trento Local Health Unit, Trento, Italy); Paolo Baldo, Sabrina Orzetti and Elisabetta Caccin (Centro di Riferimento Oncologico (CRO) di Aviano, IRCCS – Pharmacy Unit, National Cancer Institute, Aviano, Italy); Annalisa Capuano and Concetta Rafaniello (Regional Centre of Pharmacovigilance and Pharmacoepidemiology, Naples, Italy); Claudia Pagliaro, Mariangela Mercaldo, Annalisa Di Giorgio, Sonia Manna, Giuseppina Farina, Cristina Di Mauro and Michele Tari (Caserta Local Health Unit, Caserta, Italy); Ilenia De Carlo (Abruzzo Region, Pharmacovigilance Regional Centre, Italy); Ilenia De Carlo, Ilenia Senesi – Abruzzo Region, Pharmacovigilance Regional Centre, Italy; Claudia Pileggi, Caterina Palleria, Luca Gallelli, Caterina De Sarro, Chiara Verduci, Rosa Papadopoli and Giovambattista De Sarro (Department of Health Sciences, University of Catanzaro "Magna Græcia"/Regional Centre for Pharmacovigilance of Calabria, Catanzaro, Italy); Mariagrazia Morgese, Stefania Schiavone, Paolo Tucci, Maria Bove and Luigia Trabace (University of Foggia, Foggia, Italy); Francesco Lapi and Claudio Cricelli - Italian Society of General Practitioners; Giorgio Racagni (Italian Society of Pharmacology); Silvia Tonolo (Association of Patients with Rheumatic diseases); Giusi Fava, Sandro Giuffrida and Vincenza Amato (Reggio Calabria Local Health Unit, Reggio Calabria, Italy); Marco Gambera and Valentina Montresor (Ospedale Pederzoli – Hospital Pharmacy, Peschiera del Garda, Italy); Dario Mastropasqua – Croce Verde Verona, Verona, Italy. For their contribution to the Italian part of the ECVM study, as the “ilmiovaccinoCOVID19 collaborating group”. Sandra Dujmović Blažok, Morana Pavičić, Željana Margan Koletić, Ivana Ljubičić, Lara Miletić, Eva Šintić and Mario Zubec (Agency for Medicinal Products and Medical Devices of Croatia) for their contribution to the Croatian part of the ECVM study. Kenny Kestens, Dieter Cenens, Katrien Bernaert, Jamila Hamdani and Thierry Roisin for their contribution to the Belgian part of the ECVM study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The research leading to these results was conducted as part of the activities of the EU PE&PV (Pharmacoepidemiology and Pharmacovigilance) Research Network, which is a public academic partnership coordinated by the Utrecht University, The Netherlands. The project has received support from the European Medicines Agency under the Framework service contract nr EMA/2018/28/PE. This document expresses the opinions of the authors of the paper, and may not be understood or quoted as being made on behalf of or reflecting the position of the European Medicines Agency or one of its committees or working parties. In Croatia, the study was funded partially by the Agency for Medicinal Products and Medical Devices of Croatia (HALMED). In Germany, the SafeVac 2.0 study was funded by the German ministry of Health and conducted by Paul-Ehrlich-Institut the national competed authority for vaccines and biomedicinal products in Germany. The French part of the study, named EVANESCO, was partially funded by the European Medicines Agency. In France, EVANESCO was identified as a National research priority (priorité nationale de recherche) by the CAPNET, and received a complementary funding from the Ministère de la Santé et des Solidarités and the Ministère de l’Enseignement supérieur, de la Recherche et de l’Innovation. The Dutch part of the study was funded by a grant from the Dutch Ministry of Health, Welfare and Sport. The Dutch Ministry of Health, Welfare and Sport had no role in study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication. In the United Kingdom the project was funded by the European Medicines Agency through the cohort event monitoring project.
Conflicts of interest/Competing interests
Nicolas Thurin and Caroline Dureau-Pournin work for Bordeaux PharmacoEpi, an independent research platform of the Bordeaux University and its subsidiary the ADERA, which performs financially supported studies for public and private partners, in compliance with the ENCePP Code of Conduct. Miriam Sturkenboom and Sandor Schmikli work for the University Medical Center Utrecht, which performs COVID-19 vaccine trials and post-licensure vaccine effectiveness and safety studies for public and private organisations, according to ENCePP code of conduct of scientific independence. Gianluca Trifirò reported to advisory boards on topics not related to this presentation and was sponsored by the following pharmaceutical companies in the last two years: Eli Lilly, Sanofi, Amgen, Novo Nordisk, Sobi, Gilead, Celgene, Daikii Sankyo.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Availability of data and material
The datasets for this manuscript are not publicly available. Requests to access the datasets should be directed and motivated to the first author and will be granted upon reasonable request and permission from partners.
Code availability
The structured query language (SQL) statements for the data used in this article are not publicly available. Requests to access the datasets should be directed and motivated to the first author and will be granted upon reasonable request and permission from partners.
Ethical approval
The study in Belgium was approved by the Ethics Committee Research of University Hospitals Leuven. The study in Croatia received approval from Central Ethics Committee (CEC). The study in France was approved by the Comité de Protection des personnes (CPP – 21.00821.210217) and the Commission nationale de l'informatique et des libertés (CNIL – DR-2021-209) under the name EVANESCO. If a study in the Netherlands is subject to the Medical Research Involving Human Subjects Act (WMO), it must undergo a review by an accredited Medical Research Ethics Committee or the central committee on research involving human subjects (CCMO). After submission to an accredited review committee (METC Brabant), this study was deemed not to fall under the WMO act. The study in the UK was approved by the London – Brent Research Ethics Committee (REC reference: 21/HRA/2077) & obtained HRA and Health and Care Research Wales (HCRW) Approval. The study in Germany was approved by the Ethics Committee at the Regional Medical Council of Hessen. Participants in the study provided the consent electronically via the smartphone application. The study in Italy was approved by the Comitato Etico dell’Istituto Nazionale per le Malattie Infettive Lazzaro Spallanzani I.R.C.C.S. under the name “Cohort Event Monitoring of safety of COVID-19 vaccines”.
Consent to participate
Participants in the study provided written informed consent before participation to the study.
Consent for publication
Participants in the study provided a written statement of consent at the time of registration for their data to be used for the purpose of this research and publication of study results.
Author contributions
The original study protocol was designed by all authors. The queries and dataset were established by MR. Data analysis was performed by MR and FHL. The design of the manuscript was determined by MR, FHL and MS. All authors contributed to the manuscript drafting and revision. All authors approved the final version to be published and agree to be accountable for the work they performed and the methods and reporting of results.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Raethke, M., van Hunsel, F., Thurin, N.H. et al. Cohort Event Monitoring of Adverse Reactions to COVID-19 Vaccines in Seven European Countries: Pooled Results on First Dose. Drug Saf 46, 391–404 (2023). https://doi.org/10.1007/s40264-023-01281-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-023-01281-9