
Abstract
Background and objective
Cilofexor is a selective farnesoid X receptor (FXR) agonist in development for the treatment of nonalcoholic steatohepatitis and primary sclerosing cholangitis. Our objective was to evaluate potential drug-drug interactions of cilofexor as a victim and as a perpetrator.
Methods
In this Phase 1 study, healthy adult participants (n = 18–24 per each of the 6 cohorts) were administered cilofexor in combination with either perpetrators or substrates of cytochrome P-450 (CYP) enzymes and drug transporters.
Results
In total, 131 participants completed the study. As a victim, cilofexor area under the curve (AUC) was 651%, 795%, and 175% when administered following single-dose cyclosporine (600 mg; organic anion transporting polypeptide [OATP]/P-glycoprotein [P-gp]/CYP3A inhibitor), single-dose rifampin (600 mg; OATP1B1/1B3 inhibitor), and multiple-dose gemfibrozil (600 mg twice daily [BID]; CYP2C8 inhibitor), respectively, compared with the administration of cilofexor alone. Cilofexor AUC was 33% when administered following multiple-dose rifampin (600 mg; OATP/CYP/P-gp inducer). Multiple-dose voriconazole (200 mg BID; CYP3A4 inhibitor) and grapefruit juice (16 ounces; intestinal OATP inhibitor) did not affect cilofexor exposure. As a perpetrator, multiple-dose cilofexor did not affect the exposure of midazolam (2 mg; CYP3A substrate), pravastatin (40 mg; OATP substrate), or dabigatran etexilate (75 mg; intestinal P-gp substrate), but atorvastatin (10 mg; OATP/CYP3A4 substrate) AUC was 139% compared with atorvastatin administered alone.
Conclusion
Cilofexor may be coadministered with inhibitors of P-gp, CYP3A4, or CYP2C8 without the need for dose modification. Cilofexor may be coadministered with OATP, BCRP, P-gp, and/or CYP3A4 substrates—including statins—without dose modification. However, coadministration of cilofexor with strong hepatic OATP inhibitors, or with strong or moderate inducers of OATP/CYP2C8, is not recommended.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Data from this study suggest that cilofexor may be coadministered with inhibitors of P-gp, CYP3A4, or CYP2C8 without the need for dose modification. Cilofexor may be coadministered with OATP, BCRP, P-gp, and/or CYP3A4 substrates—including statins—without dose modification. However, coadministration of cilofexor with strong hepatic OATP inhibitors, or with strong or moderate inducers of OATP/CYP2C8, is not recommended. |
1 Introduction
Primary sclerosing cholangitis (PSC) and nonalcoholic steatohepatitis (NASH) represent major unmet medical needs. Primary sclerosing cholangitis is a chronic and progressive cholestatic liver disease, characterized by chronic inflammation and fibro‐obliterative destruction of intrahepatic and/or extrahepatic bile ducts, which results in progressive biliary fibrosis and cirrhosis [1]. Nonalcoholic steatohepatitis is a progressive form of nonalcoholic fatty liver disease characterized by steatosis, inflammation, hepatocyte ballooning, and hepatic fibrosis [2]. Patients with NASH have an increased risk for developing cirrhosis, liver decompensation, and hepatocellular carcinoma. Additionally, most patients suffer from comorbidities and thus are often taking multiple medications. To date, there is no approved treatment for PSC or NASH.
Cilofexor is a nonsteroidal farnesoid X receptor (FXR) agonist in clinical development for the treatment of NASH (30-mg dose in combination with other investigational agents) and PSC (100-mg dose monotherapy) [1, 3]. Cilofexor pharmacokinetics (PKs) have been characterized in healthy participants following the administration of 10- to 300-mg doses [4] and in NASH and PSC patients. Cilofexor plasma exposure showed less than dose-proportional increases in the dose range of 10 to 100 mg [4]. Cilofexor shows high protein binding in human plasma (> 99%) and is extensively metabolized, with 96.7% of the cilofexor dose eliminated in feces; 34.1% remains as intact cilofexor; and the remainder as inactive metabolites [5]. In vitro data indicate that cilofexor is a substrate of cytochrome P450 (CYP) isoforms 2C8, 2C19, and 3A, with rCYP2C8 showing the fastest rate of metabolism. Cilofexor is a substrate for human efflux transporters, P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP), and a substrate for uptake transporters, sodium taurocholate co-transporting polypeptide (NTCP) and organic anion transporting polypeptides (OATP1B1, OATP1B3, and OATP2B1). Additionally, cilofexor is an in vitro inhibitor of OATP, P-gp, and CYP3A4 (data on file, Gilead Sciences, Inc.).
The objective of this clinical study was to characterize the cilofexor drug-drug interaction (DDI) profile both as a victim and as a perpetrator in order to inform dosing recommendations for administering cilofexor with other medications that are either perpetrators or substrates of CYP enzymes and drug transporters. As a victim, the effects of cyclosporine (a mixed OATP/P-gp/multidrug resistance-associated protein 2 [MRP2]/CYP3A inhibitor) [6], rifampin (a selective OATP1B1/1B3 inhibitor [single dose] [7] and OATP/CYP3A/CYP2C8/P-gp inducer [multiple doses]) [8], voriconazole (strong CYP3A inhibitor) [9], gemfibrozil (strong CYP2C8 inhibitor/OATP1B1 inhibitor) [7], and grapefruit juice (intestinal uptake transport and CYP3A inhibitor) [10] on the exposure of cilofexor were evaluated. As a perpetrator, the effects of cilofexor on the exposure of midazolam (CYP3A substrate), atorvastatin (mixed OATP/CYP3A substrate), dabigatran etexilate (P-gp substrate), pravastatin (OATP substrate), and rosuvastatin (OATP/BCRP substrate) were evaluated [7].
2 Methods
2.1 Ethics Statement
The study protocol was reviewed and approved by an institutional review board (IRB; Schulman IRB; 4445 Lake Forest Drive, Suite 300; Cincinnati, OH 45242). The study was carried out in accordance with the Declaration of Helsinki and the International Council for Harmonisation Good Clinical Practice guidelines. All participants provided written informed consent prior to study participation.
2.2 Study Participants
Eligible participants included males and nonpregnant, nonlactating females. All participants were nonsmokers, aged between 18–45 years, had a body mass index (BMI) 19–30 kg/m2, and were required to have a normal or clinically insignificant 12-lead electrocardiogram (ECG), normal renal function, no significant medical history, and general good health at the time of screening (≤ 28 days prior to the first dose), as determined by the investigators. Exclusion criteria included pregnancy or lactation; any serious or active medical or psychiatric illness; liver disease including Gilbert’s disease; receipt of any investigational drug or device (30 days prior to first dose); use of any prescription or over-the-counter medications (except vitamins, acetaminophen, ibuprofen, and/or hormonal contraceptive medications) or herbal products within 28 days of commencing study drug dosing; a positive test result for human immunodeficiency virus 1 antibody, hepatitis B surface antigen, or hepatitis C antibody; treatment with systemic steroids, immunosuppressant therapies, or chemotherapeutic agents within 3 months prior to screening or expected to receive these agents during the study; and no current alcohol or substance abuse.
2.3 Study Design
This was a Phase 1, open-label, multicenter, multiple-cohort study in healthy participants. Following completion of screening and Day 1 assessments, eligible participants were enrolled in 1 of 6 cohorts, including 5 prespecified cohorts (Cohorts 1, 2, 3, 5, and 6) and 1 adaptive cohort (adaptive Cohort 4 [initiated based on preliminary data from Cohort 2]).
Cohort 1 assessed the effect of the administration of a single dose of cyclosporine (OATP/P-gp/CYP3A inhibitor) or rifampin (OATP1B1/1B3 inhibitor) on the exposure of a single dose of cilofexor. Participants in Cohort 1 (n = 24) were randomized and received a single dose of cilofexor 100 mg, a single dose of cyclosporine 600 mg with a single dose of cilofexor 100 mg, and a single dose of rifampin 600 mg with cilofexor 100 mg in 1 of 6 treatment sequences. Between each treatment there was a washout period on Days 2–8 and Days 10–16.
Cohort 2 assessed the effect of the administration of multiple doses of voriconazole (CYP3A4 inhibitor) or gemfibrozil (CYP2C8/OATP1B1 inhibitor) on the exposure of a single dose of cilofexor. Participants in Cohort 2 (n = 18) received a single dose of cilofexor on Day 1 followed by voriconazole 200 mg twice daily (BID) for 4 days starting on Day 9 with a single dose of cilofexor 100 mg administered on Day 9. The last dose of voriconazole was administered on Day 12 in the evening. Gemfibrozil 600 mg BID was administered for 4 days starting on Day 20 with a single dose of cilofexor administered on Day 20. The last dose of gemfibrozil was administered in the morning on Day 23. There was a 7-day washout period between each treatment.
Cohort 3 assessed the effect of the administration of multiple doses of rifampin (OATP/CYP/P-gp inducer) on the exposure of a single dose of cilofexor. Participants in Cohort 3 (n = 18) received a single 100-mg dose of cilofexor on Day 1 followed by a 7-day washout period. Rifampin 600 mg once daily was administered in the evening 1 hour before a meal for 7 days starting on Day 9. Finally, a single 100-mg dose of cilofexor was administered on Day 16 in the morning.
Cohort 4 assessed the effect of the administration of grapefruit juice (Everfresh Juice company), an intestinal OATP (and CYP3A) inhibitor, on the exposure of a single dose of cilofexor. Participants in Cohort 4 (n = 24) were randomized to 1 of 2 treatment sequences and received a single dose of cilofexor 100 mg and 16 ounces of grapefruit juice with cilofexor 100 mg. Treatments were administered on either Day 1 or Day 9 with a 7-day washout period between each treatment.
Cohort 5 assessed the effect of multiple-dose administration of cilofexor on the exposure of single doses of midazolam (CYP3A4 substrate) and atorvastatin (OATP/CYP3A4 substrate). Participants in Cohort 5 (n = 24) received a single 2-mg dose of midazolam on Day 1, followed by a single 10-mg dose of atorvastatin on Day 2, followed by a washout period on Days 3–6. Participants received cilofexor 100 mg once daily on Days 7–11 with a single dose of midazolam 2 mg on Day 7 and a single dose of atorvastatin 10 mg on Day 8.
Cohort 6 assessed the effect of multiple-dose administration of cilofexor on the exposure of single doses of dabigatran etexilate (intestinal P-gp substrate), pravastatin (OATP substrate), and rosuvastatin (OATP/BCRP substrate). Participants in Cohort 6 (n = 24) received a single dose of a cocktail of dabigatran etexilate 75 mg + pravastatin 40 mg + rosuvastatin 10 mg on Day 1 followed by a 5-day washout period. Participants received cilofexor 100 mg once daily on Days 7–10 with a single dose of the cocktail on Day 7.
A summary of cohorts and a treatment schematic are presented in Fig. 1. Participants were confined to the clinic from Day − 1 until completion of assessments on Day 21 (Cohort 1), Day 24 (Cohort 2), Day 20 (Cohort 3), Day 13 (Cohort 4), Day 12 (Cohort 5), or Day 11 (Cohort 6).

Study schematic for each cohort. ATV atorvastatin, BCRP breast cancer resistance protein, BID twice daily, CIL cilofexor, CsA cyclosporine, CYP cytochrome P450 enzyme, DE dabigatran etexilate, GFJ grapefruit juice, GFZ gemfibrozil, MDZ midazolam, OATP organic anion transporting polypeptide, P-gp P-glycoprotein, PRA pravastatin, RIF rifampin, ROS rosuvastatin, VORI voriconazole, WO washout
In all cohorts, study drugs were administered within 5 min of participants completing a standardized, moderate-fat meal. The meal was initiated 30 min prior to administration of the study drug. Participants then fasted until after collection of the PK sample at 4 hours post-dose.
2.4 Pharmacokinetic Sampling
Intensive PK sampling occurred pre-dose (< 5 min) and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16, 24, 48, 72, and 96 hours post-dose on Days 1, 9, and 17 (Cohort 1); Days 1, 9, and 20 (Cohort 2); Days 1 and 16 (Cohort 3); Days 1 and 9 (Cohort 4); Days 1, 2, 7, and 8 (Cohort 5); and Days 1 and 7 (Cohort 6).
2.5 Bioanalytical Procedures
Concentrations of cilofexor, midazolam, atorvastatin, o-hydroxyatorvastatin (o-OH-ATV), pravastatin, rosuvastatin, and dabigatran (total and free) in human plasma samples were quantified using fully validated high-performance liquid chromatography-tandem mass spectroscopy (LC-MS/MS) methods. All samples were analyzed within the time frame supported by frozen stability storage data. Lab vendor(s) analyzed plasma samples for cilofexor, midazolam, atorvastatin, o-OH-ATV, dabigatran (total and free), pravastatin, and rosuvastatin concentrations. Once the pharmacokinetic concentration data were deemed final (quality assurance performed by Covance Madison, QPS, and PPD labs), the pharmacokinetic parameters were calculated.
2.6 Pharmacokinetic Analyses
Pharmacokinetic parameters were estimated with Phoenix WinNonlin 8.2 software (Certara, LP, Princeton, NJ, USA) using standard noncompartmental methods. Samples with concentrations below the limit of quantitation of the bioanalytical assays occurring prior to the achievement of the first quantifiable concentration were assigned a concentration value of zero and at all other time points were treated as missing data in the noncompartmental analyses. Pharmacokinetic parameters included area under the plasma concentration-time curve (AUC) from time 0 to the last quantifiable concentration (AUClast), AUC extrapolated to infinity (AUCinf), maximum observed plasma concentration (Cmax), time to maximal concentration (Tmax), and terminal-phase elimination half-life (t½).
2.7 Statistical Methods
For each cohort, the selected sample size was projected to achieve at least 80% power such that the 90% CI for the geometric least-squares mean (GLSM) ratio of AUCinf, AUClast, and Cmax in test (victim drug administered with perpetrator drug) versus reference (victim drug administered alone) treatments would be within 0.70 to 1.43, if the true GLSM ratio was 1.0. For each cohort, analyte, and PK parameter, a parametric (normal theory) mixed-effects ANOVA model was fitted to the natural log-transformed values of the single-dose PK parameter under evaluation using SAS PROC MIXED. For Cohorts 1 and 4, the statistical model included treatment, sequence, and period as fixed effects and participant within sequence as a random effect. For Cohorts 2, 3, 5, and 6, the statistical model included treatment as a fixed effect and participant as a random effect. The test versus reference ratio and associated 90% CI were calculated by taking the exponential of the point estimate and the corresponding lower and upper limits, which was consistent with the two 1-sided tests approach. A lack of DDI was concluded if the GLSM ratios and corresponding 90% CIs for selected PK parameters fell within the prespecified lack of PK alteration boundaries of 70% to 143%.
2.8 Safety Assessments
Safety was monitored throughout the study. Safety was evaluated by assessment of clinical laboratory tests, ECGs, periodic physical examinations (including vital sign measurements), and documentation of adverse events (AEs). Clinical and laboratory AEs were coded using the Medical Dictionary for Regulatory Activities, Version 19.1.
3 Results
3.1 Participant Demographics
Overall, 134 participants were enrolled (24 each in Cohorts 1, 4, 5, and 6; 19 participants each in Cohorts 2 and 3); 132 participants received at least 1 dose of study drug and were included in the safety analysis set; and 131 participants (99.2%) completed the study. Two enrolled participants (1 each from Cohorts 2 and 3) did not receive study drug based on investigator discretion, and 1 participant (Cohort 4) withdrew consent on Day 1 after treatment with a single dose of cilofexor.
The mean participant age was 34 years (range, 19–45). Most participants were male (n = 78, 59.1%), White (n = 89, 67.4%), and Hispanic or Latino (n = 102, 77.3%). The mean (SD) BMI at baseline was 26.4 (2.59) kg/m2. Demographics and baseline characteristics for each cohort are presented in Table 1.
3.2 Pharmacokinetics
3.2.1 Cilofexor as a Victim of DDIs
Cilofexor mean plasma concentration versus time profiles when administered alone and with single-dose cyclosporine, grapefruit juice, single-dose rifampin, multiple-dose rifampin, multiple-dose voriconazole, and multiple-dose gemfibrozil are shown in Fig. 2. Corresponding cilofexor PK parameters, GLSM ratio, and 90% CIs are presented in Table 2 and Fig. 3.

Mean (± SD) plasma concentration of cilofexor with and without coadministration of cyclosporine (A), grapefruit juice (B), single-dose rifampin (C), multiple-dose rifampin (D), voriconazole (E), and gemfibrozil (F). For summary statistics, BLQ was treated as 0 at pre-dose and at post-dose time points. Mean concentration values that were less than or equal to the LLOQ were not displayed at post-dose time points in the plot. BLQ below limit of quantitation, LLOQ lower limit of quantitation, SD standard deviation

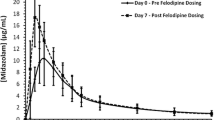
Mean (± SD) plasma concentration of probe substrates with and without coadministration of cilofexor: Midazolam (A), atorvastatin (B), free dabigatran (C), total dabigatran (D), pravastatin (E), and rosuvastatin (F). For summary statistics, BLQ was treated as 0 at pre-dose and at post-dose time points. Mean concentration values that were less than or equal to the LLOQ were not displayed at post-dose time points in the plot. BLQ below limit of quantitation, LLOQ lower limit of quantitation, SD standard deviation
Cilofexor AUC and Cmax were 651% and 512%, respectively, when cilofexor was administered with cyclosporine compared with the administration of cilofexor alone. The median cilofexor t½ was longer (approximately 2.5-fold) following administration of cilofexor with cyclosporine (Table 2, Fig. 4).

Effect of metabolizing enzymes and transporters inhibitors and inducers on cilofexor exposure. Dashed vertical lines indicate prespecified no-effect boundaries (70% to 143%). AUCinf area under the curve from time zero to infinity, CI confidence interval, Cmax maximum observed concentration, GLSMR geometric least-squares mean ratio, PK pharmacokinetics
Cilofexor AUC and Cmax were similar when cilofexor was administered with grapefruit juice compared to the administration of cilofexor alone. The 90% CIs of the GLSM ratios were contained within the strict bioequivalence 0.8 to 1.25 boundaries. Median t½ of cilofexor was similar following both treatments (Table 2).
Cilofexor AUC and Cmax were 795% and 547%, respectively, when cilofexor was administered with a single dose of rifampin compared with the administration of cilofexor alone. The median cilofexor t½ was longer (approximately 2.8-fold) following administration of cilofexor with a single rifampin dose compared with cilofexor alone (Table 2).
Cilofexor AUC and Cmax were 33% and 35%, respectively, when cilofexor was administered with multiple doses of rifampin compared with the administration of cilofexor alone. Median cilofexor t½ was slightly shorter following administration of cilofexor after multiple daily doses of rifampin (Table 2).
Cilofexor AUC was similar (the 90% CIs of the GLSM ratios were contained within the prespecified no-effect bounds [0.70–1.43]) and Cmax was 23% lower, when cilofexor was administered with multiple-dose voriconazole compared with the administration of cilofexor alone. Median cilofexor t½ was slightly longer following administration of cilofexor with voriconazole (Table 2).
Cilofexor AUC and Cmax were 175% and 139%, respectively, when cilofexor was administered with multiple-dose gemfibrozil compared with the administration of cilofexor alone. Median cilofexor t½ was slightly longer following administration of cilofexor with gemfibrozil (Table 2).
3.2.2 Cilofexor as a Perpetrator of DDIs
The changes in exposure of probe substrates with cilofexor coadministration are shown in Fig. 3. Corresponding probe substrates’ PK parameters, GLSM ratios, and 90% CIs are presented in Table 3.
Midazolam AUC and Cmax were similar when midazolam was administered with cilofexor compared to the administration of midazolam alone. The 90% CIs of the GLSM ratios were contained within the strict bioequivalence 0.8 to 1.25 boundaries. Median t½ of midazolam was similar following both treatments (Table 3).
Atorvastatin AUC was 139% and Cmax was similar when atorvastatin was administered with multiple-dose cilofexor compared to the administration of atorvastatin alone. Median atorvastatin t½ was longer following administration of atorvastatin with cilofexor versus atorvastatin alone. Administration of atorvastatin with multiple-dose cilofexor resulted in similar o-OH-atorvastatin AUC and Cmax compared to administration of the atorvastatin alone. Median o-OH-atorvastatin t½ was also longer following administration of atorvastatin with cilofexor compared with atorvastatin alone (Table 3).
Pravastatin AUC and Cmax were similar when pravastatin was administered with multiple-dose cilofexor compared to the administration of pravastatin alone. Median t½ was similar following both treatments (Table 3).
Rosuvastatin AUC and Cmax were similar when rosuvastatin was administered with multiple-dose cilofexor compared to the administration of rosuvastatin alone. Median rosuvastatin t½ was slightly longer following administration of rosuvastatin with multiple-dose cilofexor compared with rosuvastatin alone (Table 3).
Free and total dabigatran AUC and Cmax were similar when dabigatran etexilate was administered with multiple-dose cilofexor compared to the administration of dabigatran etexilate alone. Median total and free dabigatran t½ were similar following both treatments (Table 3).
3.3 Safety
Study drugs were generally well tolerated. A total of 21 of 132 participants (15.9%) experienced at least one AE, and 4 participants (3.0%) experienced an AE that was assessed by the investigator to be related to study drug. No Grade 3 or 4 AEs, serious AEs, AEs leading to premature study drug discontinuation, or deaths were reported. A summary of safety events by severity for each cohort is presented in Table 4. The most commonly reported AEs were headache (n = 5 participants, 3.8%), back pain (n = 3, 2.3%), diarrhea (n = 3, 2.3%), nasal congestion (n = 2, 1.5%), and nausea (n = 2, 1.5%). All treatment-emergent AEs (TEAEs) were Grade 1 in severity, except 1 patient who experienced a Grade 2 headache. Study drug-related AEs were reported for Cohorts 3, 4, and 6; no study drug-related AEs were reported for Cohorts 1, 2, and 5. All study drug-related AEs were Grade 1 in severity, did not require modification of study drug dose, and resolved within 1 day of onset. There were no TEAE reports of pruritus during the study. No notable changes in vital signs or clinically significant ECG abnormalities were reported.
4 Discussion
This Phase 1 study was conducted to evaluate aspects of cilofexor’s DDI profile as both a victim and as a perpetrator. Cilofexor is currently in clinical development for the treatment of NASH and PSC. Given the expected chronic use of NASH and PSC medications, characterizing their DDI potential is of great importance to ensure safe administration with other medications in the target population. Polypharmacy is a great burden among patients with diabetic nonalcoholic fatty liver disease (NAFLD), which underscores the importance of managing medications to ensure optimal treatment [11].
Observations from in vitro studies have shown that cilofexor is a substrate for P-gp, BCRP, and OATP transporters in addition to CYP3A, CYP2C8, and CYP2C9 metabolizing enzymes. Therefore, parts of this study evaluated the effect of inhibitors and inducers of these enzymes and transporters on the exposure of cilofexor. Voriconazole, a strong CYP3A inhibitor, did not impact cilofexor exposure, which indicates that CYP3A4 does not significantly contribute to cilofexor elimination. Cyclosporine, a mixed BCRP/OATP/MRP2/P-gp inhibitor and a moderate CYP3A4 inhibitor, resulted in a cilofexor AUC and Cmax of 651% and 512%, respectively, while a single dose of rifampin, a strong OAT1B1/1B3 inhibitor, resulted in a cilofexor AUC and Cmax of 795% and 547%, respectively. The more pronounced effect of rifampin on cilofexor exposure compared with cyclosporine indicates that intestinal P-gp efflux does not play a significant role in cilofexor disposition.
Grapefruit juice, an inhibitor of intestinal CYP3A and OATP1A2 [10], did not affect the exposure of cilofexor, which indicates that cilofexor is not a substrate of intestinal OATP1A2, given that in vivo perpetration to CYP3A4 does not affect the exposure of cilofexor, as discussed above. Cilofexor is a substrate of OATP2B1; the absence of an effect of grapefruit juice, an inhibitor of OAT2B1, on cilofexor exposure indicates that cilofexor and grapefruit juice do not share the same binding site on OATP2B1, similar to what is observed when pravastatin is administered with grapefruit juice [12].
Gemfibrozil, a strong CYP2C8 inhibitor, led to a cilofexor exposure AUC and Cmax of 175% and 139%, respectively, indicating that CYP2C8 is a major pathway for cilofexor metabolism. The increase in cilofexor exposure may be partially attributable to the inhibition of OATP1B1 by gemfibrozil, similar to the increase in pravastatin, an OATP1B1 substrate, exposure when it is administered with gemfibrozil [13]. This change in cilofexor exposure is not clinically relevant, based on the known exposure-safety relationship of cilofexor (data on file, Gilead Sciences, Inc.). Gemfibrozil can be administered with cilofexor without dose adjustment. Cilofexor AUC and Cmax were 33% and 36% following administration with multiple daily doses of rifampin, which is consistent with cilofexor being a substrate for hepatic OATP and CYP2C8.
Cilofexor is an in vitro inhibitor of CYP2C8, CYP3A, and CYP2C9 with half maximal inhibitory concentration (IC50) values of 2.4, 6.3, and 13.6 μM, respectively (data on file, Gilead Sciences, Inc.). Cilofexor was not a mechanism-based inhibitor of these enzymes. However, cilofexor is unlikely to cause systemic DDI in vivo through inhibition of human CYP enzymes due to high plasma protein binding (> 99%). Consistent with this, cilofexor did not alter the exposure of midazolam, a sensitive CYP3A4 substrate. It should be noted that the effect of cilofexor on midazolam was conducted because a potential CYP3A4-mediated clinical DDI was identified based on regulatory cutoff values determined by Cmax at clinical dose and IC50 value.
Cilofexor showed little to no inhibition, in vitro, of the human drug transporters P-gp, BCRP, NTCP, bile salt export pump, organic anion transporter (OAT)1, OAT3, organic cation transporter (OCT)2, multidrug and toxin extrusion protein (MATE) 1, and MATE2-K. Consistent with these findings, cilofexor did not alter the exposure of dabigatran etexilate, a probe P-gp substrate, or rosuvastatin, a BCRP/OATP substrate.
Cilofexor is an in vitro inhibitor of hepatic influx transporters OATP1B1, OATP1B3, and OATP2B1—with IC50 values of 0.68 μM, 0.41 μM, and 0.21 μM (data on file, Gilead Sciences, Inc.), respectively, which are 0.4× to 1.1× the steady-state Cmax of cilofexor following administration of cilofexor 100 mg to healthy participants under fed conditions. Cilofexor did not alter the systemic exposure of pravastatin, a sensitive OATP substrate, but caused a mild increase in AUC of atorvastatin, a mixed OATP/CYP3A substrate. The slight increase in atorvastatin exposure is not clinically relevant and does warrant atorvastatin dose modification when atorvastatin is administered with cilofexor. Also, the increase is not consistent with the fact that cilofexor did not affect the exposure of midazolam, rosuvastatin, or pravastatin. The observed slight increase in atorvastatin exposure may be related to the fact that 8 and 23 of the participants had atorvastatin concentrations below the limit of quantitation (BLQ) at the 24- and 48-hour time points, respectively, when atorvastatin was administered alone compared with 3 and 16 participants with BLQ at the same time points, respectively, when atorvastatin was administered with cilofexor.
The study findings indicate that cilofexor administration cannot be recommended with strong inhibitors of OATP, such as single-dose rifampin and cyclosporine [6, 7], or strong or moderate inducers of OATP and/or CYP2C8. Moderate inhibitors of OATP should be used with caution with cilofexor. Cilofexor can be administered with P-gp inhibitors, intestinal OATP1A2 inhibitors, CYP3A4 inhibitors, and CYP2C8 inhibitors without dose modification. Substrates of CYP3A, OATP, BCRP, or P-gp, including statins, can be administered with cilofexor without dose adjustment.
The 100-mg dose of cilofexor evaluated in this study is considered adequate to assess the magnitude of drug transporter- or metabolizing enzyme-based DDIs in the targeted patient populations. The cilofexor dose under evaluation in NASH patients is 30 mg, and in PSC patients is 100 mg [1,2,3]. The data describing cilofexor as a perpetrator can be extrapolated to the 30-mg dose, given no clinically meaningful changes in exposure of victim drugs were observed when they were administered with 100 mg cilofexor. The dose of all other interacting drugs represents their therapeutic doses. Cilofexor was administered under fed conditions to mimic the administration of cilofexor in the ongoing Phase 2/3 studies. In general, when a drug was evaluated as perpetrator in the study, it was administered in multiple doses over a duration sufficient to reach steady state and to observe the maximum inhibition/induction effect. The only exception was single-dose cyclosporine and single-dose rifampin, because as single doses, they are inhibitors of OAPT/P-gp and OATPs, respectively, while after multiple doses of rifampin, they are inducers of P-gp/OATPs and a broad CYP inducer.
All treatments were generally well tolerated. The majority of AEs were mild in nature, and no new safety signals were observed when cilofexor was administered with probe substrates and inhibitors of enzymes and transporters.
5 Conclusion
Cilofexor may be coadministered with inhibitors of intestinal OATP, P-gp, CYP3A4, or CYP2C8 without the need for dose modification. Cilofexor may be coadministered with OATP, BCRP, P-gp, and/or CYP3A4 substrates—including statins—without dose modification. However, coadministration of cilofexor with strong hepatic OATP inhibitors, or with strong or moderate inducers of OATP/CYP2C8, is not recommended.
References
Trauner M, Gulamhusein A, Hameed B, et al. The nonsteroidal farnesoid X receptor agonist cilofexor (GS-9674) improves markers of cholestasis and liver injury in patients with primary sclerosing cholangitis. Hepatology. 2019;70:788–801.
Patel K, Harrison SA, Elkhashab M, et al. Cilofexor, a nonsteroidal FXR agonist, in patients with noncirrhotic NASH: a phase 2 randomized controlled trial. Hepatology. 2020;72:58–71.
Loomba R, Noureddin M, Kowdley KV, et al. Combination therapies including cilofexor and firsocostat for bridging fibrosis and cirrhosis attributable to NASH. Hepatology. 2021;73:625–43.
Kirby B, Djedjos CS, Birkebak J, et al. Evaluation of the safety and pharmacokinetic effects of the oral, non-steroidal farnesoid X receptor agonist GS-9674 in healthy volunteers. Hepatology. 2016;63:574A.
Younis IR, Kirby BJ, Billin AN, et al. Pharmacokinetics, pharmacodynamics, safety and tolerability of cilofexor, a novel nonsteroidal farnesoid X receptor agonist, in healthy volunteers. Clin Transl Sci. 2023. https://doi.org/10.1111/cts.13469.
Yang Y, Li P, Zhang Z, Wang Z, Liu L, Liu X. Prediction of cyclosporin-mediated drug interaction using physiologically based pharmacokinetic model characterizing interplay of drug transporters and enzymes. Int J Mol Sci. 2020;21:7023.
Tornio A, Filppula AM, Niemi M, Backman JT. Clinical studies on drug-drug interactions involving metabolism and transport: methodology, pitfalls, and interpretation. Clin Pharmacol Ther. 2019;105:1345–61.
Lutz JD, Kirby BJ, Wang L, et al. Cytochrome P450 3A induction predicts P-glycoprotein induction; Part 1: establishing induction relationships using ascending dose rifampin. Clin Pharmacol Ther. 2018;104:1182–90.
Czyrski A, Resztak M, Swiderski P, Brylak J, Glowka FK. The overview on the pharmacokinetic and pharmacodynamic interactions of triazoles. Pharmaceutics. 2021;13:1961.
Bailey DG. Fruit juice inhibition of uptake transport: a new type of food-drug interaction. Br J Clin Pharmacol. 2010;70:645–55.
Patel PJ, Hayward KL, Rudra R, et al. Multimorbidity and polypharmacy in diabetic patients with NAFLD: implications for disease severity and management. Medicine (Baltimore). 2017;96: e6761.
Shirasaka Y, Mori T, Murata Y, Nakanishi T, Tamai I. Substrate- and dose-dependent drug interactions with grapefruit juice caused by multiple binding sites on OATP2B1. Pharm Res. 2014;31:2035–43.
Nakagomi-Hagihara R, Nakai D, Tokui T, Abe T, Ikeda T. Gemfibrozil and its glucuronide inhibit the hepatic uptake of pravastatin mediated by OATP1B1. Xenobiotica. 2007;37:474–86.
Acknowledgments
We extend our thanks to the study participants. This study was supported by research funding from Gilead Sciences, Inc. Writing and editorial support were provided by Helen Rodgers, PhD, of AlphaScientia, LLC, and were funded by Gilead Sciences, Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was funded by Gilead Sciences, Inc. Writing and editorial support were provided by Helen Rodgers, PhD, of AlphaScientia, LLC, San Francisco, CA, USA, and was funded by Gilead Sciences, Inc.
Conflicts of interest
EW, IY, GS, DX, TRW, AAO are employees of, and may own stock in, Gilead Sciences, Inc. CN and BJK were employees of Gilead Sciences, Inc., at the time of study and may own stock in Gilead Sciences, Inc.
Ethics approval
The study protocol was reviewed and approved by an institutional review board (IRB; Schulman IRB; 4445 Lake Forest Drive, Suite 300; Cincinnati, OH 45242).
Consent to participate
All participants provided written, informed consent prior to study participation.
Consent for publication
All authors approved this manuscript for publication.
Availability of data and material code availability
Anonymized individual participant data will be shared upon request for research purposes dependent upon the nature of the request, the merit of the proposed research, the availability of the data, and its intended use. The full data sharing policy for Gilead Sciences, Inc., can be found at https://www.gileadclinicaltrials.com/transparency-policy/.
Authors’ contributions
IY: Writing—original draft preparation; EW: Writing—original draft preparation; CN: Writing—review and editing; BK: Writing—review and editing; GS: Writing—review and editing; DX: Writing—review and editing; TW: Writing—review and editing; AAO: Writing—review and editing.
Additional information
Cara Nelson and Brian J. Kirby worked at Gilead Sciences, Inc. at the time this research was conducted.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Younis, I., Weber, E., Nelson, C. et al. Evaluation of the Potential for Cytochrome P450 and Transporter-Mediated Drug-Drug Interactions for Cilofexor, a Selective Nonsteroidal Farnesoid X Receptor (FXR) Agonist. Clin Pharmacokinet 62, 609–621 (2023). https://doi.org/10.1007/s40262-023-01214-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-023-01214-w