
Abstract
Atopic dermatitis (AD) is a chronic inflammatory disease requiring efficacious and safe long-term therapy. Several new systemic treatments have recently been approved for use in patients with moderate to severe AD. However, head-to-head comparisons have not been conducted for all the currently available treatments for AD. Multiple network meta-analyses have compared efficacy of these different therapies during the initial 16-week treatment period, but not beyond week 16. Therefore, understanding the differences in key trial design and statistical methods is essential for evaluating long-term efficacy, making cross-trial comparisons, and informing treatment decisions. This focused narrative review provides an overview of data and trial methodology to guide clinicians in evaluating longer-term efficacy and safety of currently approved systemic treatments for patients with AD. We discuss important elements of longer-term trial designs and statistical analysis strategies that should be considered based on our experience as clinical trialists. In addition, a summary of key efficacy results of published, longer-term, phase III clinical trials of US Food and Drug Administration-approved, novel systemic treatments (i.e., dupilumab, tralokinumab, abrocitinib, and upadacitinib) is provided, including the design and data handling methods used. Long-term safety considerations and differences in the time-effect and safety profiles of various medications are also noted to help inform clinical decisions for individual patients. Overall, the findings of these trials support efficacy in long-term treatment with novel systemic agents for patients with AD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Digital Features for this article can be found at https://doi.org/10.6084/m9.figshare.23796156 |
Systemic biologic agents and small molecules provide an opportunity for better disease control and improved long-term safety for patients with moderate to severe atopic dermatitis. |
It is important to understand the differences in trial design and statistical methods to compare long-term efficacy across trials and inform treatment decisions. |
The differences in efficacy and safety profiles should be discussed with patients in a shared decision-making process. |
1 Introduction
Atopic dermatitis (AD) can have a profound impact on all aspects of patients’ lives [1]. In recent years, multiple biologic agents and small molecules have been approved globally for the treatment of patients with moderate to severe AD [2,3,4,5,6]. These new systemic treatment options provide an opportunity for better disease control and improved long-term safety [2,3,4,5,6]. With more treatment options available, healthcare professionals often wrestle with determining which agents are best suited to treat their patients and have the lowest risk for adverse reactions. There have not been head-to-head comparisons for all the different AD treatments [7, 8]. Several network meta-analyses have compared the efficacy of these treatments during the initial 16-week treatment period to inform clinician decision making, but not beyond week 16 [8,9,10,11]. Because AD is a chronic disease with a complex relapsing–remitting course, longer-term assessments of efficacy are warranted [12].
Most phase III trials of treatments for patients with AD evaluate primary efficacy endpoints at week 16. Longer-term trial data extending beyond the 16-week, placebo-controlled period are more difficult to compare because trial design and analysis methods are not standardized. This further complicates risk–benefit assessments in the long term that are needed to inform treatment decisions. Silverberg et al. (2022) reviewed some key design parameters that affect results and therefore can complicate cross-trial comparisons [13]. This review provides an overview of longer-term phase III trials, evaluating US Food and Drug Administration (FDA)-approved systemic treatments for patients with AD (ie, dupilumab, tralokinumab, abrocitinib, and upadacitinib) when used with concomitant topical corticosteroids (TCS), to reflect clinical practice.
Four trials with published efficacy results for the full study population and detailed statistical analysis methods were identified for inclusion in this review: one head-to-head trial, JADE DARE (abrocitinib vs dupilumab; ClinicalTrials.gov identifier: NCT04345367) and three placebo-controlled trials, ECZTRA 3 (tralokinumab; NCT03363854), CHRONOS (dupilumab; NCT02260986), and AD UP (upadacitinib; NCT03568318) [14,15,16,17,18,19,20,21,22,23]. Although the Janus kinase (JAK) inhibitor baricitinib has been evaluated for AD treatment in multiple trials [24] and is approved for this indication in Europe and Japan [25], it has not received FDA approval [26]. We identified one longer-term study extending beyond week 16: the BREEZE-AD3 extension study (baricitinib; NCT03334435) of patients from BREEZE-AD7 (baricitinib; NCT03733301), which was conducted in a select population comprising patients who had an inadequate response to TCS and/or had failed treatment with systemic therapies within the 6 months before BREEZE-AD7 [27, 28]. Therefore, BREEZE-AD7 reflects a more refractory patient population that is markedly different from the trials reviewed here [29].
Although monotherapy trials are important to establish the efficacy of the drug, long-term use in clinical practice would include the concomitant use of topical treatment as needed. In particular, the following monotherapy studies are not reviewed here: Heads Up (upadacitinib vs dupilumab; NCT03738397) [35], BREEZE-AD3 (baricitinib; NCT03334435) [30], ECZTRA 1 and 2 (tralokinumab monotherapy; NCT03131648 and NCT03160885) [31], and SOLO-CONTINUE (dupilumab monotherapy; NCT02395133) [32]. Furthermore, we could not identify any phase III monotherapy trials with published results for the full study population (as opposed to a responder analysis) beyond week 16 for the biologics tralokinumab or dupilumab. In particular, longer-term data on maintained response at week 52 among week-16 responders are not available for the full study populations and, therefore, these studies are not reviewed here.
2 Barriers to Effective Interpretation of Longer-Term Data
2.1 Clinical Trial Design
Clinicians interpreting trial results should be aware of several key parameters of clinical trial designs that may influence outcomes. Some of the key parameters identified in the recent review by Silverberg et al. were the comparator (e.g., placebo, active control, head-to-head), the definition of and rules for rescue treatment (e.g., timing for when rescue permitted), lengths of washout periods for topical and systemic treatments (e.g., 72 h; 1, 2, or 4 weeks), and inclusion/exclusion criteria (e.g., age, disease severity) [13].
Inconsistent inclusion and exclusion criteria between trials can complicate comparative data interpretation [13]. In our experience, patients with moderate AD at baseline may be more responsive to treatment and, thus, show increased improvement, whereas those with severe AD may have more refractory disease that is less responsive to treatment [13]. Exclusion criteria can also affect safety outcomes because some trials exclude patients with more complicated medical histories who may be at risk of experiencing adverse events (AEs) [13].
The washout period refers to the time before baseline when a patient does not use medication to ensure that effects of previous treatment are no longer present prior to initiating the investigational drug [13]. A shorter washout period could affect early trial results due to an unintended enduring effect of the previous treatment. Longer washouts could allow AD to flare up, necessitating the use of rescue treatment early in the trial [13].
In trials that evaluate novel treatments in combination with TCS, use of concomitant TCS on lesional skin as required is allowed according to standard clinical practice. The type and strength of TCS used, however, may not be uniform; some trials provide patients with a list of prespecified options or with TCS in standardized kits, whereas in other trials, investigators may prescribe different TCS formulations of varying potency [13]. Trials that provide patients with TCS to use on an as-needed basis may observe more TCS use due to easier access and a reduced financial burden [13]. In addition, the use of a higher potency TCS may increase the absolute response rates compared with the use of a low-to-moderate potency TCS [13]. This can cause the placebo-adjusted response rate to go down, as the placebo arm could benefit more [13]. In longer-term trials, there is often no placebo comparator beyond week 16 for ethical reasons. This may limit the ability to estimate the efficacy of these treatments at later time points.
2.2 Analyses and Data Handling
As the length of a trial increases, so does the likelihood of missing data that could introduce bias into statistical analyses [33]. Missing data occur in almost every trial for a variety of reasons (e.g., patient withdrawal due to lack of efficacy, AEs, noncompliance). The chosen strategy for addressing the problem of missing data can greatly affect trial results. Standard approaches to handling missing data include nonresponder imputation (NRI), last observation carried forward (LOCF), analysis of as-observed data, and multiple imputation (Table 1) [34, 35]. NRI, the most conservative option, assigns a value of nonresponder to patients using rescue therapy, those who have withdrawn from the trial, and all other missing data points [33]. By assigning the worst-case response scenario to missing data, NRI analyses of investigational drug response rates can artificially estimate lower efficacy [33]. Alternatively, a less conservative option is LOCF, which inputs the last recorded value to all subsequent visits; however, this approach could overestimate the drug responses or underestimate improved longer-term responses [13, 33, 35]. As-observed data analyses evaluate only the data available at each time point and ignore missing values, and thus can overestimate treatment responses [35]. Multiple imputation considers the uncertainty of missing data by creating different plausible data sets and appropriately combining and imputing results obtained from each set [34].
2.3 Population for Analysis
The patient population used to determine efficacy is another important factor to consider. The intention-to-treat (ITT) population includes all patients randomized to each treatment arm regardless of early withdrawal, protocol deviation, or any initial response criteria requirements. If the proportions of patients who withdraw or deviate from the protocol are large, then treatment responses may be underestimated; conversely, a responder-enriched population may have the opposite bias. Placebo-treated control groups are often removed or assigned an active treatment in long-term phases of trials (e.g., after assessment of the primary endpoint), which increases the potential for bias [33].
3 Longer-Term Efficacy Data on Systemic Treatments for Atopic Dermatitis
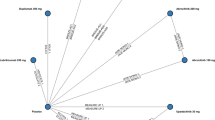
Key results from published longer-term trials of novel, FDA-approved systemic AD treatments in combination with TCS (Fig. 1) are reviewed in the following subsections, with aspects of their trial designs and data handling methods. Key binary endpoints assessed include proportions of patients achieving a score of 0 or 1 for the Investigator Global Assessment (IGA 0/1), 75% or 90% improvement in the Eczema Area and Severity Index (EASI-75 or EASI-90), and a ≥ 4-point improvement in Peak Pruritus Numerical Rating Scale (PP-NRS4).

Overview of study characteristics including trial design, key eligibility criteria, primary endpoints, safety reporting, and imputation methods. AD atopic dermatitis, AEs adverse events, BSA body surface area, EASI Eczema Area and Severity Index, HIV human immunodeficiency virus, IGA Investigator Global Assessment, MedDRA Medical Dictionary for Regulatory Activities, MI multiple imputation, NRI nonresponder imputation, PP-NRS Peak Pruritus Numerical Rating Scale, Q2W every 2 weeks, Q4W every 4 weeks, QD once daily, QW once weekly, SC subcutaneous, TB tuberculosis, TCS topical corticosteroids, vIGA validated Investigator Global Assessment
3.1 Head-to-Head Trial: JADE DARE (Abrocitinib vs Dupilumab)
JADE DARE (NCT04345367) was a 26-week, phase IIIb, randomized, double-blinded, double-dummy, active-controlled clinical trial comparing abrocitinib 200 mg once daily + TCS as needed (n = 362) with dupilumab 300 mg every 2 weeks (Q2W) + TCS as needed (n = 365) in patients aged ≥18 years with moderate to severe AD (Fig. 1) [14]. JADE DARE is the only longer-term head-to-head trial identified in our literature search that directly compared an oral JAK-1 inhibitor (abrocitinib) with an injectable biologic (dupilumab), both with concomitant TCS use. At baseline, 40% of patients had severe disease (IGA 4), and the mean EASI and PP-NRS scores were 28.1 and 7.4, respectively (Table 2) [14].
Compared with the dupilumab arm, a larger proportion of patients in the abrocitinib arm met the primary endpoints: PP-NRS4 at week 2 (48% vs 26%; p < 0.0001) and EASI-90 at week 4 (29% vs 15%; p < 0.0001) [14]. Notably, between-group differences in both endpoints decreased over time [14]. For PP-NRS4, response rates were similar at week 12 and thereafter (Figs. 2, 3, 4, 5) [14]. Proportions of patients achieving EASI-90 at week 16 were higher with abrocitinib (54% vs 42%; p = 0.0008); however, by the end of the trial at week 26, response rates were similar (55% vs 48%; not statistically significant) [14].

Proportion of patients achieving EASI-75 by study. EASI ranges from 0 to 72, with lower scores indicating less severe AD. EASI-75 is defined as an improvement of at least 75% in lesion extent and severity from baseline; thus, increases in proportions of patients achieving EASI-75 indicate greater efficacy. AD atopic dermatitis, EASI Eczema Area and Severity Index, MI multiple imputation, NRI nonresponder imputation, Q2W every 2 weeks, Q4W every 4 weeks, TCS topical corticosteroids

Proportion of patients achieving EASI-90 by study. EASI ranges from 0 to 72, with lower scores indicating less severe AD. EASI-90 is defined as an improvement of at least 90% in lesion extent and severity from baseline; thus, increases in proportions of patients achieving EASI-90 indicate greater efficacy. AD atopic dermatitis, EASI Eczema Area and Severity Index, MI multiple imputation, NRI nonresponder imputation, Q2W every 2 weeks, Q4W every 4 weeks, TCS topical corticosteroids

Proportion of patients achieving IGA 0/1 by study. The IGA score ranges from 0 to 4, with lower scores indicating less severe AD. IGA 0/1 is defined as scores of 0 or 1, indicating a clear or almost clear AD presentation; thus, increases in proportions of patients achieving IGA 0/1 indicate greater efficacy. AD atopic dermatitis, IGA Investigator Global Assessment, MI multiple imputation, NRI nonresponder imputation, Q2W every 2 weeks, Q4W every 4 weeks, TCS topical corticosteroids

Proportion of patients achieving PP-NRS4 by study. The PP-NRS score ranges from 0 to 10, with lower scores indicating less severe symptoms of itch. PP-NRS4 is defined as a ≥ 4-point improvement from baseline; thus, increases in proportions of patients achieving PP-NRS4 indicate greater efficacy. AD atopic dermatitis, MI multiple imputation, PP-NRS Peak Pruritus Numerical Rating Scale, Q2W every 2 weeks, Q4W every 4 weeks, TCS topical corticosteroids
When making cross-trial comparisons, the following elements of JADE DARE are important to consider (Fig. 1) [14, 15]:
-
Because of the lack of a placebo comparator, all patients received active therapy; therefore, any improvement from baseline might include any potential placebo response in addition to a response to study treatment.
-
Washout of TCS prior to treatment initiation was not required; rescue treatment (high-potency TCS for up to 2 weeks at a time or systemic corticosteroids for up to 10 days) to manage intolerable AD symptoms was allowed after week 4 at investigators’ discretion.
-
Concomitant TCS use was standardized by the protocol to medium- or low-potency TCS (e.g., triamcinolone acetonide 0.1% cream or fluocinolone acetonide 0.025% ointment), but not provided by the trial sponsor, and the frequency and amount used were not reported. Alternatively, topical calcineurin inhibitors (TCI) or phosphodiesterase inhibitors were allowed on areas of thin skin or if TCS were considered unsafe.
-
NRI was implemented for those patients who used rescue medication or withdrew from the trial. Other missing data were assumed to be missing at random.
3.2 Placebo-Controlled Phase III Trials
3.2.1 ECZTRA 3 (Tralokinumab)
ECZTRA 3 (NCT03363854) was a 32-week, phase III, randomized, double-blinded, placebo-controlled trial comparing tralokinumab 300 mg Q2W + TCS (n = 252) with placebo + TCS (n = 126) for an initial 16-week treatment period in patients aged ≥18 years with moderate to severe AD (Fig. 1) [17]. The initial treatment period was followed by re-randomization for an additional 16-week continuation phase; patients treated with tralokinumab achieving either primary endpoint (IGA 0/1 or EASI-75 at week 16) received either tralokinumab Q2W or every 4 weeks (Q4W), both with concomitant TCS (as needed) [16]. Patients in either arm not achieving either primary endpoint received tralokinumab Q2W, while patients who achieved a primary endpoint with placebo continued to receive placebo [16]. At baseline, 46–47% of patients had severe disease (IGA 4), and the mean EASI and mean weekly average of worst daily pruritus NRS scores were 28.8–30.4 and 7.7–7.9, respectively (Table 2) [16, 17].
At week 16, a larger proportion of patients treated with tralokinumab Q2W reached the primary endpoints compared with patients treated with placebo: IGA 0/1 (39% vs 26%; p = 0.015) and EASI-75 (56% vs 36%; p < 0.001) [16]. Similarly, a significantly greater proportion of patients treated with tralokinumab versus placebo achieved EASI-90 (33% vs 21%; p = 0.022) and mean weekly average of worst daily pruritus NRS4 (45% vs 34%; p = 0.037) [16, 17]. Prespecified continuation phase endpoints focused on the responder population and the maintained efficacy with two different dosing options [16]. A recent post-hoc analysis reported efficacy data over the entire 32-week treatment period in ECZTRA 3; this analysis included all patients initiated on tralokinumab at the start of the trial (n = 252) irrespective of their response at week 16 and tralokinumab dosing regimen (Q2W or Q4W) thereafter [17]. Response rates improved further at week 32 to 49% (IGA 0/1), 70% (EASI-75), 50% (EASI-90), and 51% (PP-NRS4, not published) (Figs. 2, 3, 4, 5) [17].
When making cross-trial comparisons, the following elements of ECZTRA 3 are important to consider (Fig. 1) [16,17,18]:
-
In the analyses beyond week 16, no placebo comparator was included and all patients received active therapy.
-
Response rates presented at week 32 are based on a post-hoc analysis conducted by pooling all patients treated with tralokinumab in the initial treatment period (n = 252), irrespective of responses at week 16 and tralokinumab dosing regimen thereafter.
-
A 2-week washout of TCS was required prior to treatment initiation; rescue treatment (higher-potency TCS: Europe class > 3; US class < 4 or systemic drugs) was allowed from the start of the trial to control intolerable AD symptoms at the investigators’ discretion.
-
This trial was the only one to standardize concomitant TCS use in the protocol to mometasone furoate 0.1% cream (Europe class 3 [potent]; US class 4 [mid-strength]), and supply this medication to patients in kit sizes of 180–200 g Q2W. Patients returned all used and unused TCS tubes, which were then weighed to determine the amount of medication used. Patients were also allowed to use low-potency TCS or TCI in areas of the body where use of the supplied TCS was inadvisable. At week 16, patients treated with tralokinumab used approximately 50% less TCS compared with patients treated with placebo; the mean use remained low (around 5–7 g/week) for patients continuing with tralokinumab treatment.
-
NRI was implemented for patients who used rescue medication or withdrew from the trial, as well as for missing data.
3.2.2 CHRONOS (Dupilumab)
CHRONOS (NCT02260986) was a 52-week, phase III, randomized, double-blinded, placebo-controlled trial comparing dupilumab 300 mg once weekly (QW; n = 319) or Q2W (n = 106) with placebo (n = 315; all arms with concomitant TCS) in patients aged ≥ 18 years with moderate to severe AD (Fig. 1) [19]. Because the FDA-approved dosing schedule for dupilumab is limited to Q2W [3], this review does not include details of the QW arm. At baseline, 47–50% of patients had severe disease (IGA 4), and the mean EASI and weekly averaged PP-NRS scores were 32.6–33.6 and 7.3–7.4, respectively (Table 2) [19, 20].
At week 16, a larger proportion of patients treated with dupilumab Q2W versus placebo reached the primary endpoints: IGA 0/1, 39% vs 12% (p < 0.0001); and EASI-75, 69% vs 23% (p < 0.0001) [19]. Similarly, a post hoc analysis showed that a significantly greater proportion of patients treated with dupilumab achieved EASI-90 (40% vs 11%; p < 0.0001) [19]. The proportion of patients achieving PP-NRS4, a secondary endpoint, was 59% with dupilumab versus 20% with placebo (p < 0.0001) [19]. At week 52, response rates with dupilumab versus placebo were maintained or slightly improved (IGA 0/1, 36% vs 13%; EASI-75, 65% vs 22%; EASI-90, 51% vs 16%; and PP-NRS4, 51% vs 13%; all p < 0.0001; Figs. 2, 3, 4, 5) [19].
For cross-trial comparisons, the following elements of CHRONOS are important to consider (Fig. 1) [19]:
-
This trial is the only one described here to include a placebo + TCS comparator group beyond week 16.
-
A 1-week washout of TCS was required prior to treatment initiation; rescue treatment (higher-potency TCS, systemic drugs, or phototherapy) to control intolerable symptoms was allowed after week 2 at the investigators’ discretion.
-
Concomitant TCS use was standardized by the protocol to medium-potency TCS (triamcinolone acetonide 0.1% cream or fluocinolone acetonide 0.025% ointment), but not provided by the trial sponsor; the amount used was not reported. Use of a low-potency TCS (hydrocortisone 1% cream) was permitted on sensitive skin areas such as the face.
-
Efficacy analyses at week 52 and time course for each trial visit included only those patients who completed the week-52 visit by the cutoff date for data submission to the FDA.
-
NRI was implemented for patients who used rescue medication or withdrew from the trial, as well as for missing data.
3.2.3 AD UP (Upadacitinib)
AD UP (NCT03568318) is an ongoing clinical trial, the first part of which was a 16-week, phase III, randomized, double-blinded, placebo-controlled trial comparing once-daily upadacitinib 15 mg (n = 300) or 30 mg (n = 297) to placebo (n = 304) (all arms with concomitant TCS use) in patients aged 12–75 years with moderate to severe AD (Fig. 1) [21]. Results from the 16-week placebo-controlled part as well as a prespecified analysis of the extension phase at week 52 have been published [21, 22]. After the initial 16-week treatment period, patients treated with upadacitinib continued treatment during the extension period for up to another 260 weeks [22]. Patients treated with placebo were rerandomized to upadacitinib, but were not included in the 52-week efficacy analysis [22]. At baseline, 52–54% of patients had severe disease (IGA 4), and the mean EASI and worst pruritus NRS responses were 29.2–30.3 and 7.1–7.4, respectively (Table 2) [21].
At week 16, a greater proportion of patients treated with upadacitinib 15 and 30 mg versus placebo reached the primary endpoints (IGA 0/1, 40% and 59% vs 11%, both p < 0.0001; and EASI-75, 65% and 77% vs 26%, both p < 0.0001) [21]. Similarly, significantly greater proportions of patients treated with upadacitinib 15 and 30 mg versus placebo achieved EASI-90 (43% and 63% vs 13%; both p < 0.0001) [21]. Proportions of patients achieving PP-NRS4 were 52% and 64% with upadacitinib 15 and 30 mg, respectively, versus 15% with placebo (both p < 0.0001) [21]. At week 52, response rates with upadacitinib 15 and 30 mg, respectively, were 34% and 45% (IGA 0/1), 51% and 69% (EASI-75), 38% and 55% (EASI-90), and 45% and 58% (PP-NRS4) (Figs. 2, 3, 4, 5) [22].
For cross-trial comparisons, the following elements of AD UP are important to consider (Fig. 1) [21, 22]:
-
There was no placebo comparator beyond week 16 and all patients received active therapy.
-
A 7-day washout of TCS was required prior to treatment initiation. Rescue treatment was allowed after week 4 if it was considered medically necessary and either of the following conditions was met:
-
The patient achieved a < 50% reduction in EASI response compared with baseline at any two consecutive scheduled visits for weeks 4–24.
-
The patient achieved a < 50% reduction at any scheduled or unscheduled visit for weeks 24–52.
-
-
Concomitant TCS was standardized by the protocol to medium-potency TCS (triamcinolone acetonide 0.1% cream or fluocinolone acetonide 0.025% ointment). Low-potency TCS (hydrocortisone 1% cream) or a topical calcineurin inhibitor was permitted for sensitive skin areas. TCS was not provided by the trial sponsor and the amount used was not reported.
-
NRI was implemented for patients who used rescue medication or withdrew from the trial and for other missing data with the exception of data that were missing due to the COVID-19 pandemic, which were handled using multiple imputation.
4 Safety Considerations
As with other data, evaluation and comparison of safety data can be influenced by a multitude of factors, including initial inclusion and exclusion criteria specified for a trial as well as trial length. Among the trials reviewed here, ECZTRA 3 presented safety data for the initial 16 weeks and the week 16–32 continuation phase [17]; JADE DARE reported safety data across the full 26-week treatment period [14]; CHRONOS reported safety data for 52 weeks [19]; and AD UP reported safety data up to 52 weeks [21, 22]. For long-term studies, it is also important to consider the frequency of AEs reported in relation to the patient years of exposure (PYE), either reported as the number of patients reporting the event per 100 PYE or the number of events per 100 PYE. In addition, differences in the ways of reporting AEs using the Medical Dictionary for Regulatory Activities (MedDRA) system can occur [36]. MedDRA categorizes AEs based on five levels: System Organ Class (SOC), High Level Group Term (HLGT), High Level Term (HLT), Preferred Term (PT), and Lowest Level Term (LLT), and depending on which level is used when reported, the same term can generate different results if reported at a PT level versus a higher-level term [36]. As an example, JADE DARE reported the conjunctivitis incidence using a higher level term that included nine PTs (allergic keratitis, conjunctival hemorrhage, conjunctivitis allergic, keratitis, noninfective conjunctivitis, ocular hyperemia, conjunctivitis, conjunctivitis bacterial, and conjunctivitis viral) [14], the CHRONOS study reported the conjunctivitis incidence, including four PTs (conjunctivitis allergic, conjunctivitis bacterial, atopic keratoconjunctivitis, and conjunctivitis) [19], and the ECZTRA 3 study reported the conjunctivitis incidence for the individual PTs [16]. Another factor to bear in mind is that there may also be differences due to use of different MedDRA versions. As an example, the most commonly reported LLT ‘common cold’ was mapped into the PT ‘viral upper respiratory tract infection’ in the MedDRA version used in the ECZTRA 3 trial, whereas it was mapped to ‘nasopharyngitis’ in the versions used for CHRONOS [17, 19]. Consequently, one of the most frequent AEs reported with tralokinumab in ECZTRA 3 was viral upper respiratory tract infection and for dupilumab in CHRONOS it was nasopharyngitis [16, 19].
Regarding treatment in clinical settings, the US prescribing information (PI), based on all trials submitted for regulatory review, can provide relevant guidance on the most important factors to consider before treatment and can highlight key differences between drug classes.
4.1 Biologics (Tralokinumab and Dupilumab)
Important exclusion criteria that were considered for the biologic trials included chronic active or acute clinically significant infections within 2–4 weeks of baseline or randomization, a history of parasitic (helminth) infection, tuberculosis infection, or known primary immunodeficiency [16, 20]. Relevant warnings or precautions listed in the PI of both biologics include a history of hypersensitivity reactions, conjunctivitis, parasitic (helminth) infections, and future need of vaccination with live vaccines; arthralgia is an additional warning for dupilumab [3, 4].
In clinical trials for tralokinumab, the most commonly reported adverse reactions (occurring in ≥ 1% of patients) were upper respiratory tract infections (mainly reported as common cold), conjunctivitis, injection-site reactions, and eosinophilia [4]. For dupilumab, the most commonly reported adverse reactions (occurring in ≥ 1% of patients) were injection-site reactions, conjunctivitis, blepharitis, oral herpes, keratitis, eye pruritus, other herpes simplex virus, dry eye, and eosinophilia [3].
4.2 JAK Inhibitors (Abrocitinib and Upadacitinib)
Important exclusion criteria that were considered for the JAK inhibitor trials included active chronic or acute infections, a history of venous thromboembolism, increased risk of an inherited coagulation disorder, a current helminth infection, tuberculosis infection, recurrent herpes zoster infections, a history of disseminated herpes zoster or herpes simplex, malignancies, and known immunodeficiency disorders [15, 23].
Boxed warnings in the PIs of the JAK inhibitors note the following: increased risk of serious infections that may lead to hospitalization or death, higher rate of all-cause mortality, malignancies, major adverse cardiovascular events, and thrombosis. Other relevant warnings or precautions include hypersensitivity reactions (upadacitinib), gastrointestinal perforations (upadacitinib), and laboratory abnormalities (upadacitinib and abrocitinib) [5, 6].
In clinical trials, the most commonly reported adverse reactions (≥ 1% of patients) for abrocitinib were nasopharyngitis, nausea, headache, herpes simplex, increased blood creatinine phosphokinase (CPK), dizziness, urinary tract infection, fatigue, acne, vomiting, impetigo, oropharyngeal pain, hypertension, influenza, gastroenteritis, contact dermatitis, upper abdominal pain, abdominal discomfort, herpes zoster, and thrombocytopenia [5]. For upadacitinib, the most commonly reported adverse reactions (≥ 1% of patients) were upper respiratory tract infections, acne, herpes simplex, headache, increased blood CPK, cough, hypersensitivity, folliculitis, nausea, abdominal pain, pyrexia, increased weight, herpes zoster, influenza, fatigue, neutropenia, myalgia, and influenza-like illness [6].
Before treatment initiation with a JAK inhibitor, recommended evaluations include a complete blood count (CBC), testing baseline hepatic function, and screening for tuberculosis and viral hepatitis [5, 6]. During treatment, CBC and lipid profiles should be monitored 4 weeks after starting treatment with either drug, 4 weeks after increasing the dose for abrocitinib, and, in our opinion, every 3−6 months thereafter [5, 6].
5 Discussion
Clinicians need meaningful comparisons of the long-term efficacy across systemic AD therapies to tailor treatment choices for their patients with moderate to severe AD to achieve optimal treatment goals. To date, a limited number of head-to-head trials have been conducted. Outside the scope of head-to-head trials, any attempt to make cross-trial comparisons is complicated by differences in trial designs and analysis methods.
To help clinicians interpret efficacy results of different trials, we highlighted key aspects of trial designs and analysis methods that should be considered due to their potential effects on results. The head-to-head JADE DARE trial’s findings support that more patients can achieve the predefined efficacy targets with a high-dose JAK inhibitor (abrocitinib 200 mg) than with a biologic (dupilumab 300 mg Q2W) during the initial treatment phase [14]. However, early between-group differences decreased over time; and at the end of the 26-week trial, proportions of patients achieving efficacy targets were similar between treatment arms [14]. These findings are consistent with the results of the placebo-controlled phase III trials, in which higher response rates were seen at week 16 with the JAK inhibitor upadacitinib (particularly at the higher dose of 30 mg) compared with the biologics dupilumab and tralokinumab but this difference diminishes over time (Figs. 2, 3, 4, 5). This result is important to consider in clinical practice for evaluating treatment response; week 16 seems too early a time point to evaluate the full benefit of the biologics.
Before initiating a new systemic therapy, it is also important to consider potential risk factors that may affect the long-term success of the treatment. From a tolerability perspective, the most frequently reported adverse drug reactions include injection-site reactions and conjunctivitis for the biologics [3, 4], and nausea and acne for the JAK inhibitors abrocitinib and upadacitinib [5, 6]. Both JAK inhibitors also have boxed warnings in their PIs for serious infections, mortality, malignancies, major adverse cardiovascular events, and thrombosis; however, observed rates of these AEs were low even during longer-term follow-up [5, 6]. Use of these medications is not recommended in combination with other JAK inhibitors, biologic immunosuppressants, or other immunomodulators, and dosage is restricted in patients aged 65 years or older (upadacitinib) or those with renal impairment (abrocitinib and upadacitinib) [5, 6]. It is important to weigh the risks and benefits of starting therapy for any patients with major cardiovascular problems, malignancies, and current or past smokers [5, 6]. Thus, patients who may be eligible for treatment with JAK inhibitors require more careful risk assessment, including periodic assessments of blood counts, liver enzymes, and lipid parameters, as well as screening for tuberculosis and viral hepatitis [5, 6].
6 Conclusion
Assessment of the efficacy of systemic AD treatments in longer-term clinical trials is challenging due to inconsistencies in trial designs and analysis methods. The trials reviewed here support the efficacy of novel systemic therapy options for long-term treatment of patients with AD. We compared different treatment options for AD, identified differences in their time-effect profiles, and contextualized the differences in study design and data handling used in each clinical trial. During initial treatment, greater proportions of patients reached the higher efficacy thresholds of EASI-90 and IGA 0/1 with JAK inhibitors than with biologics; however, during longer-term maintenance treatment, efficacy appeared to be comparable for the majority of patients. In terms of safety, different risk factors and monitoring requirements were identified for JAK inhibitors compared with biologics. These differences in efficacy and safety profiles should be discussed with patients in a shared decision-making process.
Change history
27 November 2023
A Correction to this paper has been published: https://doi.org/10.1007/s40257-023-00828-x
References
Vilsbøll AW, Anderson P, Piercy J, Milligan G, Kragh N. Extent and impact of inadequate disease control in US adults with a history of moderate to severe atopic dermatitis following introduction of new treatments. Dermatol Ther (Heidelb). 2021;11(2):475–86.
Puar N, Chovatiya R, Paller AS. New treatments in atopic dermatitis. Ann Allergy Asthma Immunol. 2021;126(1):21–31.
DUPIXENT (dupilumab) [package insert]. New York: Regeneron; 2017.
ADBRY (tralokinumab) [package insert]. New Jersey: LEO Pharma; 2021.
CIBINQO (abrocitinib) [package insert]. New York: Pfizer; 2022.
RINVOQ (upadacitinib) [package insert]. Illinois: AbbVie Biotechnology; 2019.
Langan SM, Irvine AD, Weidinger S. Atopic dermatitis. Lancet. 2020;396(10247):345–60.
Silverberg JI, Hong HC, Thyssen JP, Calimlim BM, Joshi A, Teixeira HD, et al. Comparative efficacy of targeted systemic therapies for moderate to severe atopic dermatitis without topical corticosteroids: systematic review and network meta-analysis. Dermatol Ther (Heidelb). 2022;12(5):1181–96.
Drucker AM, Morra DE, Prieto-Merino D, Ellis AG, Yiu ZZN, Rochwerg B, et al. Systemic immunomodulatory treatments for atopic dermatitis: update of a living systematic review and network meta-analysis. JAMA Dermatol. 2022;158(5):523–32.
Silverberg JI, Thyssen JP, Fahrbach K, Mickle K, Cappelleri JC, Romero W, et al. Comparative efficacy and safety of systemic therapies used in moderate-to-severe atopic dermatitis: a systematic literature review and network meta-analysis. J Eur Acad Dermatol Venereol. 2021;35(9):1797–810.
Pereyra-Rodriguez JJ, Alcantara-Luna S, Dominguez-Cruz J, Galan-Gutierrez M, Ruiz-Villaverde R, Vilar-Palomo S, et al. Short-term effectiveness and safety of biologics and small molecule drugs for moderate to severe atopic dermatitis: a systematic review and network meta-analysis. Life (Basel). 2021;11(9):927.
Chovatiya R, Silverberg JI. Evaluating the longitudinal course of atopic dermatitis: implications for clinical practice. Am J Clin Dermatol. 2022;23(4):459–68.
Silverberg JI, Simpson EL, Armstrong AW, de Bruin-Weller MS, Irvine AD, Reich K. Expert perspectives on key parameters that impact interpretation of randomized clinical trials in moderate-to-severe atopic dermatitis. Am J Clin Dermatol. 2022;23(1):1–11.
Reich K, Thyssen JP, Blauvelt A, Eyerich K, Soong W, Rice ZP, et al. Efficacy and safety of abrocitinib versus dupilumab in adults with moderate-to-severe atopic dermatitis: a randomised, double-blind, multicentre phase 3 trial. Lancet. 2022;400(10348):273–82.
Study of abrocitinib compared with dupilumab in adults with moderate to severe atopic dermatitis on background topical therapy. July 8, 2022 [cited 2022 November 10]. https://www.clinicaltrials.gov/ct2/show/NCT04345367.
Silverberg JI, Toth D, Bieber T, Alexis AF, Elewski BE, Pink AE, et al. Tralokinumab plus topical corticosteroids for the treatment of moderate-to-severe atopic dermatitis: results from the double-blind, randomized, multicentre, placebo-controlled phase III ECZTRA 3 trial. Br J Dermatol. 2021;184(3):450–63.
Silverberg JI, Adam DN, Zirwas M, Kalia S, Gutermuth J, Pinter A, et al. Tralokinumab plus topical corticosteroids as needed provides progressive and sustained efficacy in adults with moderate-to-severe atopic dermatitis over a 32-week period: an ECZTRA 3 post hoc analysis. Am J Clin Dermatol. 2022;23(4):547–59.
Tralokinumab in combination with topical corticosteroids for moderate to severe atopic dermatitis—ECZTRA 3. February 10, 2021 [cited 2022 November 10]. https://clinicaltrials.gov/ct2/show/NCT03363854.
Blauvelt A, de Bruin-Weller M, Gooderham M, Cather JC, Weisman J, Pariser D, et al. Long-term management of moderate-to-severe atopic dermatitis with dupilumab and concomitant topical corticosteroids (LIBERTY AD CHRONOS): a 1-year, randomised, double-blinded, placebo-controlled, phase 3 trial. Lancet. 2017;389(10086):2287–303.
Study to assess the efficacy and long-term safety of dupilumab (REGN668/SAR231893) in adult participants with moderate-to-severe atopic dermatitis (CHRONOS). October 17, 2017 [cited 2022 November 10]. https://clinicaltrials.gov/ct2/show/NCT02260986.
Reich K, Teixeira HD, de Bruin-Weller M, Bieber T, Soong W, Kabashima K, et al. Safety and efficacy of upadacitinib in combination with topical corticosteroids in adolescents and adults with moderate-to-severe atopic dermatitis (AD Up): results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2021;397(10290):2169–81.
Silverberg JI, de Bruin-Weller M, Bieber T, Soong W, Kabashima K, Costanzo A, et al. Upadacitinib plus topical corticosteroids in atopic dermatitis: Week 52 AD Up study results. J Allergy Clin Immunol. 2022;149(3):977–8714.
A study to evaluate upadacitinib in combination with topical corticosteroids in adolescent and adult participants with moderate to severe atopic dermatitis (AD Up). March 31, 2022 [cited 2022 November 10]. https://clinicaltrials.gov/ct2/show/NCT03568318.
Bieber T, Katoh N, Simpson EL, de Bruin-Weller M, Thaci D, Torrelo A, et al. Safety of baricitinib for the treatment of atopic dermatitis over a median of 1.6 years and up to 3.9 years of treatment: an updated integrated analysis of eight clinical trials. J Dermatolog Treat. 2023;34(1):2161812.
OLUMIANT (baricitinib) [package insert]. Madid: Lilly; 2022.
OLUMIANT (baricitinib) [package insert]. Indiana: Lilly; 2022.
Reich K, Kabashima K, Peris K, Silverberg JI, Eichenfield LF, Bieber T, et al. Efficacy and safety of baricitinib combined with topical corticosteroids for treatment of moderate to severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2020;156(12):1333–43.
Silverberg JI, Simpson EL, Thyssen JP, Werfel T, Cardillo TE, Colvin S, et al. Long-term efficacy (up to 68 weeks) of Baricitinib in combination with topical corticosteroids in adult patients with moderate-to-severe atopic dermatitis: analysis of treatment responders, partial responders and nonresponders originating from study BREEZE-AD7. J Eur Acad Dermatol Venereol. 2023;37(5):1036–45.
Blauvelt A, Teixeira HD, Simpson EL, Costanzo A, De Bruin-Weller M, Barbarot S, et al. Efficacy and safety of upadacitinib vs dupilumab in adults with moderate-to-severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2021;157(9):1047–55.
Silverberg JI, Simpson EL, Wollenberg A, Bissonnette R, Kabashima K, DeLozier AM, et al. Long-term efficacy of baricitinib in adults with moderate to severe atopic dermatitis who were treatment responders or partial responders: an extension study of 2 randomized clinical trials. JAMA Dermatol. 2021;157(6):691–9.
Wollenberg A, Blauvelt A, Guttman-Yassky E, Worm M, Lynde C, Lacour JP, et al. Tralokinumab for moderate-to-severe atopic dermatitis: results from two 52-week, randomized, double-blind, multicentre, placebo-controlled phase III trials (ECZTRA 1 and ECZTRA 2). Br J Dermatol. 2021;184(3):437–49.
Worm M, Simpson EL, Thaci D, Bissonnette R, Lacour JP, Beissert S, et al. Efficacy and safety of multiple dupilumab dose regimens after initial successful treatment in patients with atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2020;156(2):131.
Papp KA, Fonjallaz P, Casset-Semanaz F, Krueger JG, Wittkowski KM. Analytical approaches to reporting long-term clinical trial data. Curr Med Res Opin. 2008;24(7):2001–8.
Sterne JA, White IR, Carlin JB, Spratt M, Royston P, Kenward MG, et al. Multiple imputation for missing data in epidemiological and clinical research: potential and pitfalls. BMJ. 2009;29(338): b2393.
Langley RG, Reich K. The interpretation of long-term trials of biologic treatments for psoriasis: trial designs and the choices of statistical analyses affect ability to compare outcomes across trials. Br J Dermatol. 2013;169(6):1198–206.
MedDRA. [cited 2023 March 16]; MedDRA Hierarchy. https://www.meddra.org/how-to-use/basics/hierarchy.
Acknowledgements
We thank the patients and their families of each included trial for participating in their relevant trials. Medical writing support was provided by Dhara Sardhara, MD, and Raya Mahbuba, MSc, on behalf of Oxford PharmaGenesis Inc., Newtown, PA, USA, and was funded by LEO Pharma.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Not applicable.
Conflict of interest
J.I. Silverberg: Dr Silverberg has received honoraria as a consultant and/or advisory board member for AbbVie, Alamar, Aldena, Amgen, AObiome, Arcutis, Arena, Asana, Aslan, BioMX, Biosion, Bodewell, Boehringer Ingelheim, Bristol Meyers Squibb, Cara, Castle Biosciences, Celgene, Connect Biopharma, Dermavant, Dermira, Dermtech, Eli Lilly, Galderma, GlaxoSmithKline, Incyte, Kiniksa, LEO Pharma, Menlo, Novartis, Optum, Pfizer, RAPT, Recludix, Regeneron, Sanofi-Genzyme, Shaperon, Union, and UpToDate; speaker fees for AbbVie, Eli Lilly, LEO Pharma, Pfizer, Regeneron, and Sanofi-Genzyme; and his institution received grants from Galderma, Incyte, and Pfizer. A. Armstrong: Dr Armstrong has served as a consultant for and received honoraria from AbbVie, Almirall, Arcutis, ASLAN, Beiersdorf, BI, BMS, EPI, Incyte, LEO Pharma, UCB, Janssen, Lilly, Nimbus, Novartis, Ortho Dermatologics, Sun, Dermavant, Dermira, Sanofi, Regeneron, and Pfizer; she has participated in advisory boards for Boehringer Ingelheim and Parexel. A. Blauvelt: Dr Blauvelt has served as a speaker (received honoraria) for AbbVie, Bristol Myers Squibb, Eli Lilly and Company, Pfizer, Regeneron, and Sanofi; he has served as a scientific adviser (received honoraria) for AbbVie, Abcentra, Aclaris, Affibody, Aligos, Almirall, Alumis, Amgen, Anaptysbio, Arcutis, Arena, Aslan, Athenex, Bluefin Biomedicine, Boehringer Ingelheim, Bristol Myers Squibb, Cara Therapeutics, Dermavant, EcoR1, Eli Lilly and Company, Escient, Evelo, Evommune, Forte, Galderma, HighlightII Pharma, Incyte, InnoventBio, Janssen, Landos, LEO Pharma, Merck, Novartis, Pfizer, Rani, Rapt, Regeneron, Sanofi Genzyme, Spherix Global Insights, Sun Pharma, TLL Pharmaceutical, TrialSpark, UCB Pharma, Union, Vibliome, and Xencor; and has acted as a clinical study investigator (institution has received clinical study funds) for AbbVie, Acelyrin, Allakos, Almirall, Alumis, Amgen, Arcutis, Athenex, Boehringer Ingelheim, Bristol Myers Squibb, Concert, Dermavant, Eli Lilly and Company, Evelo, Evommune, Galderma, Incyte, Janssen, LEO Pharma, Merck, Novartis, Pfizer, Regeneron, Sun Pharma, UCB Pharma, and Ventyx. K. Reich: Professor Reich has served as advisor and/or paid speaker for and/or participated in clinical trials sponsored by AbbVie, Almirall, Amgen, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Forward Pharma, Gilead, Galderma, Janssen-Cilag, Kyowa Kirin, LEO Pharma, Eli Lilly and Company, Medac, Novartis, Ocean Pharma, Pfizer, Sanofi, UCB; Professor Reich is co-founder of Moonlake Immunotherapeutics.
Ethics approval
Not applicable.
Patient consent to participate/publish
Not applicable.
Availability of data and material
The authors confirm that the data supporting the findings of this study are available within the article and/or its supplementary materials. These data were derived from the cited references in the public domain.
Code availability
Not applicable.
Author contributions
JI, AA, AB, and KR substantially contributed to the conception of this article, including interpreting the relevant literature, and have critically revised it for intellectual content. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Silverberg, J.I., Armstrong, A., Blauvelt, A. et al. Assessment of Efficacy and Safety Outcomes Beyond Week 16 in Clinical Trials of Systemic Agents Used for the Treatment of Moderate to Severe Atopic Dermatitis in Combination with Topical Corticosteroids. Am J Clin Dermatol 24, 913–925 (2023). https://doi.org/10.1007/s40257-023-00809-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40257-023-00809-0