
Abstract
Introduction
An unsafe injection practice is one of the major contributors to new hepatitis C virus (HCV) infections; thus, people who inject drugs are a key population to prioritize to achieve HCV elimination. The introduction of highly effective and well-tolerated pangenotypic direct-acting antivirals, including glecaprevir/pibrentasvir (GLE/PIB), has revolutionized the HCV treatment landscape. Glecaprevir is a weak cytochrome P450 3A4 (CYP3A4) inhibitor, so there is the potential for drug–drug interactions (DDIs) with some opioids metabolized by CYP3A4, such as fentanyl. This study estimated the impact of GLE/PIB on the pharmacokinetics of intravenous fentanyl by building a physiologically based pharmacokinetic (PBPK) model.
Methods
A PBPK model was developed for intravenous fentanyl by incorporating published information on fentanyl metabolism, distribution, and elimination in healthy individuals. Three clinical DDI studies were used to verify DDIs within the fentanyl PBPK model. This model was integrated with a previously developed GLE/PIB PBPK model. After model validation, DDI simulations were conducted by coadministering GLE 300 mg + PIB 120 mg with a single dose of intravenous fentanyl (0.5 µg/kg).
Results
The predicted maximum plasma concentration ratio between GLE/PIB + fentanyl and fentanyl alone was 1.00, and the predicted area under the curve ratio was 1.04, suggesting an increase of only 4% in fentanyl exposure.
Conclusion
The administration of a therapeutic dose of GLE/PIB has very little effect on the pharmacokinetics of intravenous fentanyl. This negligible increase would not be expected to increase the risk of fentanyl overdose beyond the inherent risks related to the amount and purity of the fentanyl received during recreational use.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out this study? |
In recent years, the opioid epidemic in the United States has resulted in increasing numbers of overdose deaths, particularly due to synthetic opioids like fentanyl. |
This study evaluated the potential drug interaction between glecaprevir/pibrentasvir and intravenous fentanyl using a physiologically-based pharmacokinetic model. |
What was learned from this study? |
The model predicted a negligible effect of glecaprevir/pibrentasvir coadministration on the pharmacokinetics of intravenous fentanyl. |
To achieve HCV elimination, HCV-infected people who use drugs must be prioritized for treatment, preferably in an integrated care setting that addresses their addiction challenges, which should not be withheld despite their ongoing drug use. |
Introduction
Illicit injection drug use (IDU) has been linked to many transmissible diseases, including viral infections such as hepatitis C. In 2019, approximately 58 million people worldwide were estimated to be living with chronic hepatitis C virus (HCV) infection, with 1.5 million new infections each year [1]. Globally, an unsafe injection practice is the primary route for new HCV infections, with an estimated 67–95% of new infections being due to IDU in Europe and North America between 2018 and 2019 [1,2,3]. People who inject drugs (PWID) are a key population who need to be prioritized in order to achieve HCV elimination [1]. Several clinical trials as well as real-world evidence confirm the efficacy of pangenotypic direct-acting antivirals (DAAs) in this population, making HCV elimination among PWID by 2030 a distinct possibility [4, 5].
Fentanyl is a synthetic opioid agonist used as an anesthetic adjunct or commonly administered in the hospital setting for postsurgical or other treatment-related pain management [6,7,8]. Most commonly, it is administered through intravenous (IV), intramuscular, and transdermal routes, but there are also oral formulations in the form of buccal tablets [6, 7, 9, 10]. It is 50–100 times more potent than morphine, with a shorter onset and duration of action, and a narrow therapeutic index. Because of this, there is a potential for severe adverse events (AEs) if the dose is not carefully adjusted and monitored [7, 10, 11]. In addition to being used as a prescribed therapeutic, fentanyl is commonly used as a recreational drug. In this setting, without any pharmaceutical drug quality assessments, and without appropriate monitoring and dose-adjustment strategies, the risk and severity of AEs are obviously higher than when administered in a controlled setting. Recreational fentanyl use presents the highest risk of unintended drug–drug interactions (DDIs) due to the uncontrolled dose and frequency of administration [12, 13]. This is particularly relevant as there were approximately 71,000 overdose deaths involving synthetic opioids in 2021 in the United States [10, 14, 15]. Fentanyl use is often unintentional, as it may be mixed with other drugs purchased for recreational use without the user’s knowledge [16, 17].
Fentanyl is believed to be primarily metabolized in the liver by the enzyme cytochrome P450 3A4 (CYP3A4) into the inactive metabolite norfentanyl [18, 19], and as such, CYP3A4 inhibitors may increase fentanyl exposure. The US Food and Drug Administration (FDA) and European Medicine Agency (EMA) classify CYP3A inhibitors as strong, medium, or weak based on their pharmacokinetic (PK) effects on CYP3A substrates [20,21,22]. Strong CYP3A4 inhibitors such as ritonavir and moderate inhibitors such as voriconazole and fluconazole have been shown to increase the IV fentanyl plasma concentration by 174%, 40%, and 26%, respectively [23, 24]. In contrast, midazolam, which is known to be exclusively metabolized by CYP3A4, presents observed exposure increases of 1450% and 250% in the presence of ritonavir and fluconazole [25, 26]. This discrepancy in the change in CYP3A4 substrate exposure highlights that fentanyl metabolism is only partially mediated by CYP3A4, an observation supported by studies finding that fentanyl clearance was only 8% mediated by CYP3A4 relative to midazolam, and that itraconazole had no statistically significant effects on the PK of fentanyl [27, 28]. The relative contributions of various metabolic and drug transport pathways to the total in vivo elimination of fentanyl are also influenced by the route of administration (oral/transdermal/IV) [19].
The combination of glecaprevir/pibrentasvir (GLE/PIB) is a pangenotypic antiviral regimen approved to treat chronic HCV infection [29, 30]. GLE is classified as a weak CYP3A4 inhibitor due to a 27% increase in midazolam exposure, which may be a concern for narrow therapeutic index drugs that rely on CYP3A4 for clearance [29, 30]. As clinical studies of the DDIs between DAAs and fentanyl are lacking, physiologically based pharmacokinetic (PBPK) models can be a useful tool for simulating real-world scenarios of DDIs which cannot be captured in clinical trials. By combining a computational model representing the whole human body with physiological parameters, drug properties, and information from previous clinical trials, a PBPK model attempts to synthesize available information on the PK processes of a certain drug in order to predict its PK in untested scenarios [31,32,33,34]. Several PK clinical studies have confirmed the predictive value of PBPK models [35,36,37].
The aim of this study was to build and verify a PBPK model to simulate and estimate the impact of the weak CYP3A4 inhibitor GLE, in combination with PIB, on the PK of IV fentanyl.
Methods
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Software
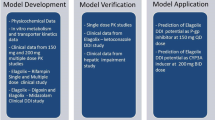
PBPK models integrate three different types of parameters: (i) drug-specific properties (of fentanyl in this study), (ii) human physiological information (pertaining to healthy Caucasian individuals in this study), and (iii) clinical trial design specifics related to the dose, regimen, and length of the trials being simulated (Supplemental Fig. S1). The PBPK model for fentanyl was developed using the Simcyp® simulator (version 20; Certara, Princeton, NJ), and the built-in “Sim-Healthy” population library was used to conduct virtual clinical trial simulations with 100 virtual healthy individuals. The Simcyp® software incorporates population-level physiological characteristics for healthy volunteers and includes known population distributions of physiological parameters such as tissue volumes, blood flow rates, metabolizing enzyme abundances, and plasma protein levels [38].
Fentanyl PBPK Model Development
The PBPK model was developed for IV fentanyl (chosen because this leads to higher bioavailability compared with other routes of administration) and its major metabolite, norfentanyl, by incorporating published information on fentanyl metabolism, distribution, and elimination in humans [39,40,41]. The model simulated IV fentanyl administration and therefore oral absorption was not considered.
As metabolic DDIs were the focus of this study, a minimal PBPK model, based on model parsimony, was selected to model in vivo distribution. Because IV fentanyl is primarily metabolized in the liver, the model included the liver as a separate metabolizing compartment (to account for the expression and abundance of metabolizing enzymes and the associated inter-individual variability), while all the other tissues were combined into a central systemic compartment and a single adjusting compartment (SAC). The structure of the minimal PBPK model for fentanyl was presented in [39], and information from previously published PBPK models for fentanyl were utilized in this analysis [39,40,41].
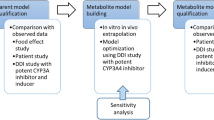
The PBPK model was calibrated and verified using clinical PK data on IV fentanyl with a dose range of 1.5–100 µg/kg from six clinical studies (Table 1) [42,43,44,45,46,47]. The initial value of the volume of distribution of the central compartment (Vss) for fentanyl was 3.857 L/kg, based on Ji et al. [39]. However, comparison of the predicted PK profile of IV fentanyl with observed clinical data suggested that a single compartment distribution model was not sufficient. As a result, a SAC was added to the model. By varying the parameter value for Vss, the volume of SAC (VSAC), and the blood flow to the SAC (QSAC), final values were selected for these parameters which resulted in the best agreement of the predicted PK profile with the clinical observations (Supplemental Table S1).
Although fentanyl has been shown to be a substrate of P-gp transporter in vitro, this transporter was not included in the PBPK model as it has been shown that it does not contribute significantly to the disposition of IV fentanyl [48].
Kovar et al. developed a PBPK model for IV fentanyl which estimated fentanyl to be predominantly metabolized through hepatic CYP3A4 activity (33%) [40] to norfentanyl, but noted that other non-CYP3A pathways of metabolism existed. Between 4 and 10% of the unchanged fentanyl is removed through urinary excretion [19, 40, 43], and 7% by systemic elimination through to renal elimination [40]. In the PBPK model by Kovar et al., the authors included active efflux of fentanyl by P-gp in the liver [40]. P-gp transporters are expressed in the canalicular membrane of hepatocytes and act by actively pumping xenobiotics into the bile canaliculi, which drain into the bile duct, leading to biliary elimination. However, the authors did not quantify the percentage contribution of this elimination pathway. In our study, the model by Kovar et al. was retrospectively utilized by shutting down the P-gp transporter pathway, which resulted in a 30% increase in the AUC of IV fentanyl. Based on this, a contribution of 30% was assigned to the biliary elimination of fentanyl. Therefore, the remaining 24% was assigned to nonspecific elimination pathways, which account for the unknown metabolites which have been identified for fentanyl [18]. Ultimately, the contributions of the different pathways were confirmed by comparing the model predictions with multiple DDI studies.
Fentanyl PBPK Model DDI Verification
To verify the ability of the PBPK model to simulate DDIs, three clinical DDI studies in which fentanyl was coadministered with the CYP3A4 inhibitors fluconazole [24], ketoconazole [43], and ritonavir [23] were used. For this purpose, the maximum plasma concentration (Cmax) and the area under the curve (AUC) ratios (with and without inhibitors) predicted by the model were compared to the parameters observed in the clinical studies to ensure they were within the prespecified acceptance criteria. The model simulation designs followed the clinical study designs with respect to doses, regimens, and length of coadministration. For these simulations, inbuilt inhibitor PBPK models for fluconazole, ketoconazole, and ritonavir (available within Simcyp® V20) were used without any change.
GLE/PIB PBPK Model Development
The PBPK models for GLE/PIB used in this study were previously developed, reported, and published [49]. Model predictions were used to evaluate the risk of DDIs between fentanyl and GLE/PIB (Fig. 1). In vitro studies and a clinical DDI study suggested that GLE is a weak CYP3A4 inhibitor. In the clinical DDI study, the coadministration of 300 mg GLE with the CYP3A4 substrate midazolam increased the AUC of oral midazolam by only 27% [29, 30].

The Advanced Dissolution, Absorption and Metabolism (ADAM) module was used within Simcyp® V20 to model the gastric dissolution and absorption of GLE and PIB. P-glycoprotein (P‐gp) and breast cancer resistant protein (BCRP) transporters were included for the efflux of PIB in the gut as well as in the canalicular efflux in the liver, whereas for GLE, only the P‐gp transporters were included. OATP1B1/1B3 transporters are important for the active hepatic uptake of GLE, which undergoes metabolic elimination by the enzyme CYP3A4. The observed nonlinear PKs were captured by the saturation of P‐gp and OATP 1B1/1B3 transporters for GLE and by the saturation of P‐gp and BCRP transporters for PIB. Because renal elimination plays a minimal role in the total clearance of GLE and PIB, it was not included for either compound. PIB is not known to have significant metabolic elimination, and hence additional elimination was assigned as the nonspecific clearance of PIB.
PBPK Model Application: Fentanyl-GLE/PIB DDI Prediction
Following the validation of the PBPK model of fentanyl and GLE/PIB, the simulation of DDIs between fentanyl and GLE/PIB was conducted. Coadministration of 300 mg GLE + 120 mg PIB along with a single dose of IV fentanyl (0.5 µg/kg) was simulated. The dose of IV fentanyl was administered at day 11, after 11 daily doses of GLE/PIB. This was done to make sure that GLE/PIB plasma concentrations were at a steady state when fentanyl was administered in order to elicit the maximum possible DDI.
Results
Model PK Calibration and Verification
The PBPK model prediction, based on an average of 100 virtual individuals given an IV bolus dose of fentanyl, shows good agreement with the observed values for patients receiving one or two doses of IV fentanyl 100 µg/kg (Supplemental Fig. S2a) [42], 10 µg/kg (Supplemental Fig. S2b) [44], or a single dose of IV fentanyl 5 µg/kg (Supplemental Fig. S2c) [43]. The PBPK model also gives a good prediction for the increase in the systemic concentration of norfentanyl after a single dose of IV fentanyl 5 µg/kg (Supplemental Fig. S2d) [43]. The model was then verified against previously published data: patients receiving a single bolus injection of fentanyl 0.5 mg (Supplemental Fig. S2e) [45]; patients receiving IV 75 µg/kg (Supplemental Fig. S2f) [46]; and patients receiving either 100 µg, 200 µg, or 300 µg (Supplemental Fig. S2g–i) [47].
Model DDI Verification
Fentanyl
Combined PBPK models of fentanyl and norfentanyl were able to capture observed DDIs with CYP3A inhibitors such as fluconazole, ketoconazole, and ritonavir within the acceptance criteria (Table 2) [23, 24, 43].
GLE/PIB
Two separate models were developed using the physicochemical and absorption, distribution, metabolism, and excretion (ADME) properties of GLE and PIB to capture the observed nonlinear PK for both drugs. The GLE/PIB PBPK model described all the clinical data for GLE and PIB both individually and as a combination. The model-simulated Cmax and AUC ratios were within the variable prediction margin acceptance criteria, with about 20% exceptions [50]. DDIs between GLE/PIB and fentanyl were simulated using the same clinical study design used to study DDIs with midazolam (Fig. 2) [51]. The DDI for the coadministration of fentanyl and GLE/PIB was validated using the DDI results for midazolam at two different doses of GLE and PIB, which showed that the predicted numbers are within the acceptance criteria (Fig. 2) [51]. At a 700 mg dose of GLE and 160 mg of PIB, the predicted Cmax ratio between GLE/PIB + midazolam (1 mg single dose) and midazolam alone was 1.33 and the predicted AUC ratio was 2.71, whereas at lower GLE/PIB doses (300 mg/120 mg), the predicted Cmax ratio was 1.05 and the predicted AUC ratio was 1.20.

Summary of PBPK model verification for the effect of GLE/PIB on the CYP3A4 substrate midazolam [51, 52]. PBPK model simulations were conducted with 100 virtual individuals and following the study design of the clinical study with respect to dose and regimen [51]. Geometric mean ratios of Cmax and AUC are presented. *Acceptance criteria based on Guest et al. (2011) [50]. AUC area under the curve; Cmax maximum plasma concentration; GLE glecaprevir; PBPK physiologically based pharmacokinetic; PIB pibrentasvir
Model Predictions for GLE/PIB and Fentanyl
Based on model predictions, at the 300 mg dose of GLE and 120 mg of PIB, the predicted Cmax ratio between GLE/PIB + fentanyl and fentanyl alone was 1.00, and the predicted AUC ratio was 1.04 (1.04–1.05), an increase of 4% in fentanyl exposure (Fig. 3).

PBPK model simulations for GLE/PIB + fentanyl. aModel predicted geometric mean ratios for 100 virtual individuals. Geometric mean ratios and 90% CIs (in parentheses) are shown. AUC area under the curve; Cmax maximum plasma concentration; CI confidence interval; GLE glecaprevir; IV intravenous; PBPK physiologically based pharmacokinetic; PIB pibrentasvir
Discussion
This study evaluated the potential interaction between GLE and fentanyl as a CYP3A4 substrate. A fentanyl PBPK model was developed, clinically verified, and used to predict the impact of the weak CYP3A4 inhibitor GLE on the PK of the synthetic opioid fentanyl. The PBPK simulation demonstrated that the coadministration of GLE/PIB at the therapeutic dose of 300 mg/120 mg has a negligible effect (a 4% increase) on IV fentanyl PK parameters at 0.5 µg/kg, providing reassurance to health care providers (HCPs) regarding potential PK interactions between GLE/PIB and street-purchased fentanyl.
The negligible predicted increase of IV fentanyl PK when coadministered with GLE was in line with our expectation based on published literature. Compared with fentanyl alone, the concomitant use of fentanyl with strong (ritonavir) or moderate (fluconazole) CYP3A4 inhibitors can increase the AUC0–∞ of IV fentanyl by 174% and 23%, respectively [23, 24]. Since GLE is a weak CYP3A4 inhibitor which increased the plasma AUC of the CYP3A4 substrate midazolam by only 27%, its interaction with fentanyl was expected to be less pronounced compared with the effect of moderate or strong CYP3A4 inhibitors [29, 30]. The metabolism of fentanyl is not fully understood, and a few lines of evidence have suggested that some unknown metabolic pathways may be involved, reducing the metabolic action of CYP3A4 to just 8% [27, 43]. One study showed that the strong CYP3A4 inhibitor itraconazole did not affect the PK of IV fentanyl [28].
The focus of this study was on IV fentanyl because this method of administration provides the highest bioavailability and the highest risk for potentially clinically significant interactions compared with other administration routes [6, 7, 9, 10]. Additionally, because the most common route of HCV transmission is through IDU, the choice of the IV route is likely to be most applicable in the real world to HCV-infected PWID who may be considered for DAA therapy [1]. PWID are a priority population for diagnosis, linkage to care, and treatment if the World Health Organization’s goal of HCV elimination as a public health concern by 2030 is to be reached [1]. Therefore, it is important to understand whether there is any potential for DDIs between DAAs, such as GLE/PIB and fentanyl, because fentanyl use is frequently unintentional due to its addition to other recreational drugs without the user’s knowledge [16, 17]. Although available data on DDIs between DAAs and opioids are limited, findings from real-world studies that included people who were actively using drugs suggest that the coadministration of GLE/PIB with recreational drugs has no apparent impact on safety, and PK studies show that DDIs between DAAs and opioids are unlikely [4, 5, 53,54,55]. The risk of fentanyl-related overdose may be more likely associated with the amount and purity of the fentanyl exposure as opposed to the effect of any concomitant DAA therapy, including GLE/PIB [56]. A recent analysis showed that, of the ~ 58,000 fentanyl AEs recorded in the FDA Adverse Event Reporting System (FAERS), only a small proportion (0.07%; n = 40) have been reported in association with any concomitant DAA [57]. In the United States, synthetic opioids such as fentanyl account for 66% of the 107,622 drug overdoses recorded in 2021, suggesting that drug overdose remains the main cause of death related to synthetic opioids such as fentanyl [14].
Limitations of this study include the assumption that P-gp transporters do not play an essential role in the disposition of IV fentanyl, and were therefore not incorporated into the model simulation [48, 58]. In a clinical study conducted separately with oral and IV fentanyl administered along with the sensitive P-gp inhibitor quinidine, P-gp appeared to have less effect on the brain access of fentanyl [48]. A clinical DDI study investigating the role of the OATP1B1 uptake transporter pathway in fentanyl disposition found that a 600 mg single dose of rifampin (OATP1B1 inhibitor) reduced IV fentanyl elimination by only 20% [58]. As rifampin at single doses is also a P-gp inhibitor, this 20% reduction in fentanyl elimination suggests a minimal role of P-gp in IV fentanyl clearance [58, 59]. Additionally, in a study comparing common mu-opioid analgesics in wild-type and P-gp knockout mice, the increase in fentanyl levels in mice brain was modest and comparable to the 20% reduction in fentanyl elimination observed after IV administration [60].
This model captured the contribution of biliary elimination to the in vivo disposition of IV fentanyl. Biliary elimination is a combination of passive diffusion at the canalicular membrane of the liver as well as active efflux mediated by transporters such as P-gp. Based on available information in the literature about the minimal role of hepatic P-gp in the elimination of fentanyl, biliary elimination was assumed to be solely due to passive diffusion [58, 59]. Additionally, the model did not evaluate other routes of administration (e.g., oral or transdermal), which may make varying contributions via CYP3A4 due to other metabolic and drug-transport mechanisms. It is expected that any interaction associated with other routes of administration would be less significant than those associated with IV fentanyl, providing reassurance of the applicability of this model to all methods of recreational fentanyl use. Lastly, it must be noted that the PBPK model simulations performed in this work were based on a virtual Caucasian population. However, fentanyl PK has not been shown to be affected by ethnicity [61]. Population PK analysis of fentanyl PK in patients has not found race or ethnicity to be a significant covariate [61]. The DDI explored here between GLE and fentanyl is due to the CYP3A4 enzyme, which is not known to have significant polymorphic variants in different populations [61].
Conclusion
These findings indicate that the administration of the therapeutic dose of 300 mg/120 mg GLE/PIB would have very little effect on the PK of IV fentanyl, suggesting that HCPs have greater evidence for reassurance regarding DDIs between GLE/PIB and fentanyl. These data also support the recommendations from the American Association for the Study of Liver Diseases, the Infectious Diseases Society of America, the American Society of Addiction Medicine, and the European Association for the Study of the Liver, which state that all patients with hepatitis C should be treated with any of the available pangenotypic DAA regimens, regardless of ongoing IDU [62,63,64]. Breaking down the barriers and facilitating HCV treatment uptake among this traditionally underserved population is fundamental to achieving HCV elimination, and all available and indicated DAA options should be considered to maximize the likelihood of successful treatment initiation.
References
World Health Organization (WHO). Global progress report on HIV, viral hepatitis and sexually transmitted infections, 2021. Accountability for the global health sector strategies 2016–2021: actions for impact. 2021. https://www.who.int/publications/i/item/9789240027077. Accessed March 2023.
Trickey A, Fraser H, Lim AG, Peacock A, Colledge S, Walker JG, et al. The contribution of injection drug use to hepatitis C virus transmission globally, regionally, and at country level: a modelling study. Lancet Gastroenterol Hepatol. 2019;4(6):435–44.
Jin F, Dore GJ, Matthews G, Luhmann N, Macdonald V, Bajis S, et al. Prevalence and incidence of hepatitis C virus infection in men who have sex with men: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol. 2021;6(1):39–56.
Conway B, Truong D, Yamamoto L, Yung R, Sharma S. Use of glecaprevir/pibrentasvir (G/P) for the treatment of HCV infection among fentanyl users: an interim analysis of the GRAND PLAN study. J Hepatol. 2022;77:S553.
Moga T, Nallapeta N, Martinez A, editors. Variable adherence to HCV treatment among people who inject drugs treated with 8 versus 12 weeks of antiviral therapy resulted in high rates of SVR12 and reinfection rate was low. In: The Liver Meeting American Association for The Study Of Liver Disease, AASLD, 2022 Nov 4–8; Washington, DC, USA.
Datapharm Ltd. Electronic Medicine Compendium (EMC). Matrifen 75 micrograms/hour Transdermal patch. 2023. https://www.medicines.org.uk/emc/product/10272/. Accessed March 2023.
Datapharm Ltd. Electronic Medicine Compendium (EMC). Fentanyl 50 microgram/ml injection. 2022. https://www.medicines.org.uk/emc/product/2852/smpc#gref. Accessed March 2023.
Centers for Disease Control and Prevention (CDC). Fentanyl. 2022. https://www.cdc.gov/opioids/basics/fentanyl.html. Accessed March 2023.
Ramos-Matos CF, Bistas KG, Lopez-Ojeda W. Fentanyl. Treasure Island: StatPearls Publishing; 2022.
Stanley TH. The fentanyl story. J Pain. 2014;15(12):1215–26.
Kornick CA, Santiago-Palma J, Moryl N, Payne R, Obbens EA. Benefit-risk assessment of transdermal fentanyl for the treatment of chronic pain. Drug Saf. 2003;26(13):951–73.
Drug Enforcement Administration. National drug threat assessment. 2019. https://www.dea.gov/sites/default/files/2020-01/2019-NDTA-final-01-14-2020_Low_Web-DIR-007-20_2019.pdf. Accessed 2023 March.
Centers for Disease Control and Prevention (CDC). The facts about fentanyl. 2022. https://www.cdc.gov/stopoverdose/fentanyl/index.html. Accessed Mar 2023.
Centers for Disease Control and Prevention (CDC). U.S. overdose deaths in 2021 increased half as much as in 2020—but are still up 15%. 2022. https://www.cdc.gov/nchs/pressroom/nchs_press_releases/2022/202205.htm. Accessed March 2023.
Ciccarone D. Fentanyl in the US heroin supply: a rapidly changing risk environment. Int J Drug Policy. 2017. https://doi.org/10.1016/j.drugpo.2017.06.010.
Griswold MK, Chai PR, Krotulski AJ, Friscia M, Chapman B, Boyer EW, et al. Self-identification of nonpharmaceutical fentanyl exposure following heroin overdose. Clin Toxicol. 2018;56(1):37–42.
Mars SG, Rosenblum D, Ciccarone D. Illicit fentanyls in the opioid street market: desired or imposed? Addiction. 2019;114(5):774–80.
Feierman DE, Lasker JM. Metabolism of fentanyl, a synthetic opioid analgesic, by human liver microsomes. Role of CYP3A4. Drug Metab Dispos. 1996;24(9):932–9.
Kuip EJ, Zandvliet ML, Koolen SL, Mathijssen RH, van der Rijt CC. A review of factors explaining variability in fentanyl pharmacokinetics; focus on implications for cancer patients. Br J Clin Pharmacol. 2017;83(2):294–313.
US Food and Drug Administration (FDA). Drug development and drug interactions: table of substrates, inhibitors and inducers. 2022. https://www.fda.gov/drugs/developmentapprovalprocess/developmentresources/druginteractionslabeling/ucm093664.htm. Accessed March 2023.
Hachad H, Ragueneau-Majlessi I, Levy RH. A useful tool for drug interaction evaluation: the University of Washington Metabolism and Transport Drug Interaction Database. Hum Genom. 2010;5(1):1–12.
European Medicines Agency (EMA). Guideline on the investigation of drug interactions. 2012. https://www.ema.europa.eu/documents/scientific-guideline/guideline-investigation-drug-interactions_en.pdf. Accessed March 2023.
Olkkola KT, Palkama VJ, Neuvonen PJ. Ritonavir’s role in reducing fentanyl clearance and prolonging its half-life. Anesthesiology. 1999;91(3):681–5.
Saari TI, Laine K, Neuvonen M, Neuvonen PJ, Olkkola KT. Effect of voriconazole and fluconazole on the pharmacokinetics of intravenous fentanyl. Eur J Clin Pharmacol. 2007;64(1):25–30.
Olkkola KT, Ahonen J, Neuvonen PJ. The effect of the systemic antimycotics, itraconazole and fluconazole, on the pharmacokinetics and pharmacodynamics of intravenous and oral midazolam. Anesth Analg. 1996;82(3):511–6.
Greenblatt DJ, Harmatz JS. Ritonavir is the best alternative to ketoconazole as an index inhibitor of cytochrome P450–3A in drug–drug interaction studies. Br J Clin Pharmacol. 2015;80(3):342–50.
Geist MJ, Ziesenitz VC, Bardenheuer HJ, Burhenne J, Skopp G, Mikus G. Minor contribution of cytochrome P450 3A activity on fentanyl exposure in palliative care cancer patients. Sci Rep. 2019;9(1):1–6.
Palkama VJ, Neuvonen PJ, Olkkola KT. The CYP 3A4 inhibitor itraconazole has no effect on the pharmacokinetics of iv fentanyl. Br J Anaesth. 1998;81(4):598–600.
AbbVie. Mavyret: US package insert. 2021. https://www.rxabbvie.com/pdf/mavyret_pi.pdf. Accessed March 2023.
AbbVie. Maviret: EU summary of product characteristics. 2021. https://www.ema.europa.eu/en/documents/product-information/maviret-epar-product-information_en.pdf. Accessed March 2023.
Cordes H, Thiel C, Baier V, Blank LM, Kuepfer L. Integration of genome-scale metabolic networks into whole-body PBPK models shows phenotype-specific cases of drug-induced metabolic perturbation. NPJ Syst Biol Appl. 2018;4(1):1–11.
Einolf HJ, Lin W, Won CS, Wang L, Gu H, Chun DY, et al. Physiologically based pharmacokinetic model predictions of panobinostat (LBH589) as a victim and perpetrator of drug-drug interactions. Drug Metab Dispos. 2017;45(12):1304–16.
Freise KJ, Shebley M, Salem AH. Quantitative prediction of the effect of CYP3A inhibitors and inducers on venetoclax pharmacokinetics using a physiologically based pharmacokinetic model. J Clin Pharmacol. 2017;57(6):796–804.
Zhang X, Yang Y, Grimstein M, Fan J, Grillo JA, Huang SM, et al. Application of PBPK modeling and simulation for regulatory decision making and its impact on US prescribing information: an update on the 2018–2019 submissions to the US FDA’s Office of Clinical Pharmacology. J Clin Pharmacol. 2020;60:S160–78.
Zhou W, Johnson T, Xu H, Cheung S, Bui K, Li J, et al. Predictive performance of physiologically based pharmacokinetic and population pharmacokinetic modeling of renally cleared drugs in children. CPT Pharmacomet Syst Pharmacol. 2016;5(9):475–83.
Willmann S, Thelen K, Kubitza D, Lensing AW, Frede M, Coboeken K, et al. Pharmacokinetics of rivaroxaban in children using physiologically based and population pharmacokinetic modelling: an EINSTEIN-Jr phase I study. Thromb J. 2018;16(1):1–12.
Tistaert C, Heimbach T, Xia B, Parrott N, Samant TS, Kesisoglou F. Food effect projections via physiologically based pharmacokinetic modeling: predictive case studies. J Pharm Sci. 2019;108(1):592–602.
Jamei M, Marciniak S, Feng K, Barnett A, Tucker G, Rostami-Hodjegan A. The Simcyp® population-based ADME simulator. Expert Opin Drug Metab Toxicol. 2009;5(2):211–23.
Ji B, Liu S, Xue Y, He X, Man VH, Xie X-Q, et al. Prediction of drug–drug interactions between opioids and overdosed benzodiazepines using physiologically based pharmacokinetic (PBPK) modeling and simulation. Drugs R&D. 2019;19(3):297–305.
Kovar L, Weber A, Zemlin M, Kohl Y, Bals R, Meibohm B, et al. Physiologically-based pharmacokinetic (PBPK) modeling providing insights into fentanyl pharmacokinetics in adults and pediatric patients. Pharmaceutics. 2020;12(10):908.
Shankaran H, Adeshina F, Teeguarden JG. Physiologically-based pharmacokinetic model for fentanyl in support of the development of provisional advisory levels. Toxicol Appl Pharmacol. 2013;273(3):464–76.
Björkman S. Reduction and lumping of physiologically based pharmacokinetic models: prediction of the disposition of fentanyl and pethidine in humans by successively simplified models. J Pharmacokinet Pharmacodyn. 2003;30(4):285–307.
Ziesenitz VC, König SK, Mahlke NS, Skopp G, Haefeli WE, Mikus G. Pharmacokinetic interaction of intravenous fentanyl with ketoconazole. J Clin Pharmacol. 2015;55(6):708–17.
Bentley JB, Borel JD, Nenad RE Jr, Gillespie TJ. Age and fentanyl pharmacokinetics. Anesth Analg. 1982;61(12):968–71.
Stoeckel H, Schüttler J, Magnussen H, Hengstmann J. Plasma fentanyl concentrations and the occurrence of respiratory depression in volunteers. Br J Anaesth. 1982;54(10):1087–95.
Christrup LL, Foster D, Popper LD, Troen T, Upton R. Pharmacokinetics, efficacy, and tolerability of fentanyl following intranasal versus intravenous administration in adults undergoing third-molar extraction: a randomized, double-blind, double-dummy, two-way, crossover study. Clin Ther. 2008;30(3):469–81.
Mather LE, Woodhouse A, Ward ME, Farr SJ, Rubsamen RA, Eltherington LG. Pulmonary administration of aerosolised fentanyl: pharmacokinetic analysis of systemic delivery. Br J Clin Pharmacol. 1998;46(1):37–43.
Kharasch ED, Hoffer C, Altuntas TG, Whittington D. Quinidine as a probe for the role of p-glycoprotein in the intestinal absorption and clinical effects of fentanyl. J Clin Pharmacol. 2004;44(3):224–33.
Mukherjee D, Kosloski M, Liu W, Shebley M. Physiologically based pharmacokinetic modeling of glecaprevir and pibrentasvir as a combination: mechanistic modeling of non-linear pharmacokinetics. Poster presentation (PII-099) at ASCPT Annual Meeting, 2018 Mar 21–24; Orlando, FL, USA.
Guest EJ, Aarons L, Houston JB, Rostami-Hodjegan A, Galetin A. Critique of the two-fold measure of prediction success for ratios: application for the assessment of drug-drug interactions. Drug Metab Dispos. 2011;39(2):170–3.
Center for Drug Evaluation and Research. Clinical pharmacology and biopharmaceutics review(s). 2017. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/209394orig1s000clinpharmr.pdf.
Kosloski M, Dutta S, Pugatch D, Li H, Mensa F, Kort J, et al. ABT-493 and ABT-530 combination demonstrated minimal potential for CYP-mediated drug-drug interactions. J Hepatol. 2016;2(64):S405.
Persico M, Aglitti A, Milella M, Coppola C, Messina V, Claar E, et al. Real-life glecaprevir/pibrentasvir in a large cohort of patients with hepatitis C virus infection: the MISTRAL study. Liver Int. 2019;39(10):1852–9.
Cornberg M, Ahumada A, Aghemo A, Andreoni M, Bhagat A, Butrymowicz I, et al. Safety and effectiveness using 8 weeks of glecaprevir/pibrentasvir in HCV-infected treatment-naïve patients with compensated cirrhosis: the CREST Study. Adv Ther. 2022. https://doi.org/10.1007/s12325-022-02158-6.
Ing Lorenzini K, Girardin F. Direct-acting antiviral interactions with opioids, alcohol or illicit drugs of abuse in HCV-infected patients. Liver Int. 2020;40(1):32–44.
US International Trade Commission. Deadly high-purity fentanyl from China is entering the U.S. through e-commerce channels. 2019. https://www.usitc.gov/publications/332/executive_briefings/ebot_george_serletis_fentanyl_from_china_pdf.pdf.
Martinez AKT, Dylla DE, et al. Reported safety of HCV direct-acting antivirals with opioids: 2017 to 2021. Poster presentation (FRI411) at EASL. J Hepatol. 2022. https://doi.org/10.1016/S0168-8278(22)01509-4.
Ziesenitz VC, König SK, Mahlke N, Jantos R, Skopp G, Weiss J, et al. Fentanyl pharmacokinetics is not dependent on hepatic uptake by organic anion-transporting polypeptide 1b1 in human beings. Basic Clin Pharmacol Toxicol. 2013;113(1):43–8.
Reitman M, Chu X, Cai X, Yabut J, Venkatasubramanian R, Zajic S, et al. Rifampin’s acute inhibitory and chronic inductive drug interactions: experimental and model-based approaches to drug–drug interaction trial design. Clin Pharmacol Ther. 2011;89(2):234–42.
Mercer LS, Coop A. Opioid analgesics and P-glycoprotein efflux transporters: a potential systems-level contribution to analgesic tolerance. Curr Top Med Chem. 2011;11(9):1157–64.
Smith HS. Opioid metabolism. Mayo Clin Proc. 2009;84(7):613–24.
American Association for the Study of Liver Diseases (AASLD), Infectious Disease Society of America (IDSA). HCV guidance: recommendations for testing, managing, and treating hepatitis C. 2022. https://www.hcvguidelines.org/.
American Society of Addiction Medicine. Public policy statement on hepatitis C infection. 2017. https://www.asam.org/docs/default-source/public-policy-statements/pdff5b01a9472bc604ca5b7ff000030b21a.pdf?sfvrsn=0#:~:text=Current%20guidelines%2C%20therefore%2C%20by%20the,I%2C%20Level%20A).%E2%80%9D.
Pawlotsky J-M, Negro F, Aghemo A, Berenguer M, Dalgard O, Dusheiko G, et al. EASL recommendations on treatment of hepatitis C: final update of the series. J Hepatol. 2020;73(5):1170–218.
Acknowledgements
Funding
AbbVie sponsored the study, contributed to its design, and participated in the collection, analysis, and interpretation of the data and in the writing, reviewing, and approval of the abstract. AbbVie funded the journal’s Rapid Service fees associated with journal publication. All authors had access to all relevant data and participated in the writing, review, and approval of the manuscript. No honoraria or payments were made for authorship.
Medical Writing, Editorial, and Other Assistance
The authors would like to express their gratitude to the patients who participated in this study and their families, as well as the study investigators and coordinators of the study. Glecaprevir was identified by AbbVie and Enanta. Medical writing support was provided by Marta Rossi, PhD, and Laura Whiteley, PhD, of Fishawack Health Inc, and funded by AbbVie.
Authorship
All named authors meet the International Committee of Medical Journal Editors criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
Conceptualization: Dimitri Semizarov, Jatinder Kaur, Nael M. Mostafa, Dwaipayan Mukherjee, Michelle Collins.Data curation: Dwaipayan Mukherjee.Formal analysis: Dwaipayan Mukherjee.Investigation: Dwaipayan Mukherjee.Methodology: Dwaipayan Mukherjee, Nael M. Mostafa.Validation: Dwaipayan Mukherjee, Nael M. Mostafa.Writing—original draft: Dwaipayan Mukherjee, Michelle Collins, Douglas E. Dylla, Jatinder Kaur, Dimitri Semizarov, Anthony Martinez, Brian Conway, Tipu Khan, Nael M. Mostafa.Writing—review & editing: Dwaipayan Mukherjee, Michelle Collins, Douglas E. Dylla, Jatinder Kaur, Dimitri Semizarov, Anthony Martinez, Brian Conway, Tipu Khan, Nael M. Mostafa.
Disclosures
Dwaipayan Mukherjee, Michelle Collins, Douglas E. Dylla, Jatinder Kaur, Dimitri Semizarov, and Nael M. Mostafa are employees of AbbVie and may hold AbbVie stock/options.Anthony Martinez: speaking fees from AbbVie, Gilead, Salix, and Eisai; consulting fees from AbbVie and Gilead; research grants from AbbVie, Gilead, Intercept, and Allergan.Brian Conway: grants, honoraria, and research support from AbbVie Inc., Gilead Sciences, Merck & Co., and ViiV Healthcare.Tipu Khan: speaker, consultant, and advisory board member for AbbVie, Alkermes, and Indivior.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial-level data (analysis data sets), as well as other information (e.g., protocols, clinical study reports, or analysis plans), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent, scientific research, and will be provided following the review and approval of a research proposal and statistical analysis plan (SAP) and the execution of a data sharing agreement (DSA). Data requests can be submitted at any time after approval in the US and Europe and after acceptance of this manuscript for publication. The data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Mukherjee, D., Collins, M., Dylla, D.E. et al. Assessment of Drug–Drug Interaction Risk Between Intravenous Fentanyl and the Glecaprevir/Pibrentasvir Combination Regimen in Hepatitis C Patients Using Physiologically Based Pharmacokinetic Modeling and Simulations. Infect Dis Ther 12, 2057–2070 (2023). https://doi.org/10.1007/s40121-023-00830-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40121-023-00830-0