
Abstract
Background and Objective
Combined acetaminophen and ibuprofen are common antipyretic and analgesic drugs. Formulation and feeding affect drug absorption. Drug clearance has a nonlinear relationship with total body weight. The covariate effect of fat mass on acetaminophen and ibuprofen pharmacokinetics remains unexplored. This study sought to quantify acetaminophen and ibuprofen pharmacokinetics with intravenous, tablet, sachet and oral suspension formulations in fed and fasted states.
Methods
Pooled time–concentration data for acetaminophen and ibuprofen were available from fasting and fed healthy adults. Data from intravenous, tablet, sachet and suspension formulations were analysed using nonlinear mixed-effects models. Body composition was considered as a covariate on clearances and volumes of distribution (Vd). Size metrics investigated were total body weight, fat and fat-free mass. Theory-based allometry was used to scale pharmacokinetic parameters to a 70 kg individual. A factor on absorption half-life and lag time quantified delays due to feeding for oral formulations. Pharmacokinetic-pharmacodynamic simulations were used to explore the time courses of pain response for acetaminophen and ibuprofen for each formulation.
Results
Pooled data included 116 individuals (18–49 years, 49–116 kg) with 6095 acetaminophen and 6046 ibuprofen concentrations available for analysis. A two-compartment pharmacokinetic model with first-order elimination described disposition for both drugs. Normal fat mass was the best covariate to describe acetaminophen clearance (CL), with a factor for fat contribution (FFATCL) of 0.816. Acetaminophen volume of distribution was described using total body weight. Normal fat mass was the best covariate to describe ibuprofen clearance (FFATCL = 0.863) and volume of distribution: (FFATV = 0.718). Clearance and central volume of distribution were 24.0 L/h/70 kg and 43.5 L/h/70 kg for acetaminophen. Ibuprofen clearance and central volume of distribution were 3.79 L/h/70 kg and 10.5 L/h/70 kg. Bioavailability and absorption half-life were 86% and 12 min for acetaminophen and 94% and 27 min for ibuprofen. Absorption lag times were 5.3 min and 6.7 min for acetaminophen and ibuprofen, respectively. Feeding increased both absorption half-life and absorption lag time when compared to the tablet formulation under fasting conditions. Feeding had the most pronounced effect on the lag time associated with tablet formulation for both drugs. Time to a pain score reduction of 2 points (visual analogue score, 0–10) differed by only 5–10 min across all formulations for acetaminophen and ibuprofen.
Conclusion
Fat mass was an important covariate to describe acetaminophen and ibuprofen pharmacokinetics. The absorption half-lives of acetaminophen and ibuprofen were increased in fed states. The delay in absorption, quantified by a lag time, was protracted for both drugs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The pharmacokinetic parameters clearance and volume of distribution can be used to calculate drug dose. Fat mass has an impact on acetaminophen clearance and on both clearance and volume of distribution for ibuprofen. |
Feeding decreases the rate and extent of absorption for acetaminophen and ibuprofen tablet, suspension and sachet formulations. |
The acetaminophen and ibuprofen formulations investigated had no meaningful clinical impact on the reduction of pain score. |
1 Introduction
Acetaminophen and ibuprofen are first-line analgesics that can be used to control pain in children and adults [1, 2]. Both drugs have analgesic and antipyretic properties. The primary site of acetaminophen action is debated but its effects are thought to be exerted via inhibition of prostaglandin synthesis [3]. Prostaglandin H2 synthase (PGHS) is the enzyme responsible for the metabolism of arachidonic acid to the unstable prostaglandin H2. The two major forms of this enzyme are the constitutive PGHS-1 and the inducible PGHS-2. PGHS comprises two sites: a cyclooxygenase (COX) site and a peroxidase (POX) site. The conversion of arachidonic acid to prostaglandin G2 is dependent on a tyrosine-385 radical at the COX site. Acetaminophen acts as a reducing co-substrate on the POX site and lessens the amount of tyrosine-385 radical at the COX site. Nonsteroidal anti-inflammatory drugs (NSAIDs) act by reducing prostaglandin biosynthesis through inhibition at the COX site of PGHS [4]. Although both drugs act through inhibition of prostaglandin H2 synthase (PGHS), acetaminophen lacks the anti-inflammatory effects of the NSAIDs [5].
Multimodal analgesia is desirable for pain control. Combining drugs with distinct mechanisms of action reduces the dose requirements of individual analgesics and spares the use of opioids [6]. Due to different mechanisms of action, acetaminophen and ibuprofen can be combined without increasing the incidence of adverse effects such as hepatic [7] or gastrointestinal [8] toxicity. The pharmacodynamics of combined acetaminophen and ibuprofen have been described using response surface methodology [2, 9].
In clinical trials, combined acetaminophen and ibuprofen (500 mg acetaminophen + 150 mg ibuprofen and 325 mg acetaminophen + 97.5 mg ibuprofen) was effective in the control of pain following tooth extraction [1, 10]. The combination of acetaminophen and ibuprofen is superior to analgesia from either drug alone, with participants receiving combination therapy less likely to require additional analgesia for breakthrough pain [1, 11, 12].
Pharmaceutical formulations of combined acetaminophen and ibuprofen have been developed: a sachet formulation to facilitate faster absorption than a tablet, an intravenous formulation, and an oral suspension formulation indicated for use in children 2–12 years old. Feeding can affect the rate and extent of drug absorption due to physiological changes in the gastrointestinal tract (e.g. altered gastric emptying rate or pH) or by physical interactions with food and drug molecules [13].
The pharmacokinetic parameters clearance (CL) and volume of distribution (Vd), which are used to determine dose, may be different in the obese compared to the non-obese [14,15,16,17,18,19]. The composition of the body can be considered in its simplest form as consisting of fat mass and fat-free mass. Fat-free mass can be predicted in adults using age, sex, weight and height [20]. Combinations of components may be used to predict both CL and Vd. One body composition descriptor may be suitable for the loading dose and another required for the maintenance dose because Vd and CL, which are responsible for these different dosing phases, are independent of each other. Covariate effects of body composition on acetaminophen pharmacokinetics have been described [21] but remain unexplored for ibuprofen.
The objective of this study was to describe acetaminophen and ibuprofen pharmacokinetics with intravenous, tablet, sachet and oral suspension formulations. Specifically, we aimed to quantify the influence of feeding in healthy adult volunteers and to assess body composition as a covariate to describe differences between individuals in acetaminophen and ibuprofen pharmacokinetics.
2 Methods
Pooled time–concentration data for acetaminophen and ibuprofen were available from fasting and fed healthy adults [22]. Data from intravenous, tablet, sachet and suspension formulations were analysed using nonlinear mixed-effects models.
2.1 Data Sources
These studies were approved by the Jordan Food and Drug Administration and were registered with the Australian New Zealand Clinical Trial Registry (AFT-MXIV-01—ACTRN12614000809639; AFT-MXIV-06—ACTRN12615001208594; AFT-MX-14a- ACTRN12616000418471; AFT-MX-14b—ACTRN12616000419460).
2.1.1 Study 1 (MXIV-01)
This study was a phase I, single-centre, single-dose, open-label, randomised, five-way crossover trial in 30 healthy adult participants. Treatments were: Maxigesic® IV (an intravenous formulation of acetaminophen 1000 mg + ibuprofen 300 mg in a 100 mL infusion); intravenous acetaminophen 1000 mg in a 100 mL infusion; intravenous ibuprofen 300 mg in a 100 mL infusion; half-dose Maxigesic® IV (acetaminophen 500 mg + ibuprofen 150 mg in a 100 mL infusion); and two Maxigesic® film-coated tablets (each tablet containing acetaminophen 500 mg + ibuprofen 150 mg). Subjects were fasted for at least 10 h prior to drug administration. Intravenous formulations were infused over 15 min into an intravenous cannula. Eighteen blood samples were drawn after intravenous administration. Samples were obtained before the first dose was administered, on completion of the 15 min of infusion, and at 5, 10, 15, 20, 30, and 45 min and 1, 1.25, 1.5, 2, 3, 4, 6, 8, 10 and 12 h after completion of infusion. Sixteen blood samples were drawn after tablet administration. Samples were obtained before the first dose was administered and at 5, 10, 20, 30, and 45 min and 1, 1.25, 1.50, 2, 3, 4, 6, 8, 10 and 12 h after the study drug administration.
2.1.2 Study 2 (MXIV-06)
This study was a phase I, single-centre, single-dose, open-label, randomised, four-way crossover trial in 30 healthy adult participants. Treatments were: Maxigesic® IV (an intravenous formulation of acetaminophen 1000 mg + ibuprofen 300 mg in a 100 mL infusion); Ofirmev® (intravenous acetaminophen 1000 mg in a 100 mL infusion); Caldolor® (intravenous ibuprofen 400 mg in a 4 mL infusion); and three Maxigesic® film-coated tablets (each tablet containing acetaminophen 325 mg + ibuprofen 97.5 mg). Subjects were fasted for at least 10 h prior to drug administration. Maxigesic® IV and Ofirmev® were administered as an intravenous infusion over 15 min into an indwelling intravenous cannula. Caldolor® was administered as an intravenous infusion over 30 min into an indwelling intravenous cannula. Eighteen blood samples were drawn after intravenous administration. Samples were obtained before the first dose was administered, on completion of the intravenous infusion, and at 5, 10, 15, 20, 30, and 45 min and 1, 1.25, 1.5, 2, 3, 4, 6, 8, 10 and 12 h after completion of infusion. Sixteen blood samples were drawn after tablet administration. Samples were obtained before the first dose was administered and at 5, 10, 20, 30 and 45 min and 1.00, 1.25, 1.50, 2, 3, 4, 6, 8, 10 and 12 h after drug administration.
2.1.3 Study 3 and Study 4 (MX-14a and 14b)
Each of these studies was a single-centre, open-label, randomised, single-dose study with a four-way crossover design. There were 28 healthy adult participants enrolled in each study. The same drug treatments were administered under fasting conditions in Study 3 and fed conditions in Study 4. Treatments were: Maxigesic® Oral Suspension (an oral liquid formulation of 1000 mg acetaminophen + 300 mg ibuprofen in 31.25 mL [160 mg acetaminophen + 48 mg ibuprofen/5 mL]); Maxigesic® Sachet (a powder formulation of 1000 mg acetaminophen + 300 mg ibuprofen dissolved in 200 mL water); Maxigesic®, two film-coated tablets (total dose 1000 mg acetaminophen + 300 mg ibuprofen); and Maxigesic® 325, three film-coated tablets (total dose 975 mg acetaminophen + 292.5 mg ibuprofen). There was a washout period of 3 days between each two consecutive study drug administrations. Fourteen blood samples were drawn from each individual. Samples were obtained pre-dose and at 5, 15, 30, and 45 min and 1, 1.25, 1.50, 2, 3, 6, 8, 10 and 12 h post dose in each period. The total volume of blood drawn did not exceed 42 mL.
2.2 Drug Assay
Acetaminophen and ibuprofen concentrations were measured with liquid chromatography–tandem mass spectrometry (LC/MS/MS). A set of nine acetaminophen nonzero (0.05–20 mg/L) calibration standards were prepared. Quality control samples were prepared at concentrations of 0.15, 2.5, 10 and 15 mg/L. A set of ten ibuprofen nonzero calibration standards between 0.05 and 35 mg/L were prepared. Quantity control samples were prepared at 0.15, 1.25, 12.5, 17.5 and 27 mg/L.
Calibration standards and quality controls for both drugs were prepared in human plasma. Acceptable limits of precision and accuracy for calibration standards were ± 15% and ± 20% at the lower limit of quantification (LLOQ) for acetaminophen and paracetamol, respectively. The LLOQ for both assays was 0.05 mg/L. Data below LLOQ were treated as zero. Correlation coefficients were equal to or greater than 0.99.
2.3 Pharmacokinetic Modelling
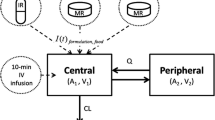
Acetaminophen and ibuprofen pharmacokinetics were investigated using one- and two-compartment models with first-order elimination and first-order absorption with a lag time for formulations given orally. Population analyses were performed using nonlinear mixed effects models (NONMEM 7.5, ICON Development Solutions, Hanover, MD, USA). Pharmacokinetic models were parameterised in terms of elimination clearance (CL) from the central compartment, intercompartment clearances (Q2), central and peripheral volumes of distribution (e.g. V1, V2), absorption half-life (T1/2 ABS) and lag time (TLAG). An additional factor (FFED) was used to quantify the effect of feeding on T1/2 ABS and TLAG for each drug formulation. Size differences were described using the following equation [23, 24]:
where Fsize is the fractional difference in size when scaled using allometry and WT is the weight of the ith individual. The allometric theory-based exponent (EXP) was fixed at ¾ for clearance parameters and 1 for distribution volumes [25].
The covariate effect of body composition on the pharmacokinetic parameters CL and Vd was investigated using total body mass (TBM, kg), fat-free mass (FFM, kg) and normal fat mass (NFM, kg). FFM was predicted using
where WHSmax is the maximum FFM for any given height (HT, m) and WHS50 is the TBM value when FFM is half of WHSmax. For men, WHSmax is 42.92 kg/m2 and WHS50 is 30.93 kg/m2, and for women WHSmax is 37.99 kg/m2 and WHS50 is 35.98 kg/m2 [20].
NFM is a size descriptor based on allometric theory describing contributions from fat mass and FFM. NFM is FFM plus a component of fat mass which can be described using the parameter Ffat (Eq. 3) [26]. The effect of FFM on CL and V was assessed by fixing Ffat at zero (i.e. considering the effect of FFM alone) (Eq. 4).
Allometric body mass can be determined using a standard value for NFM known as NFMSTD. The standardised value for NFM can be defined using a FFM of 56.1 kg, which is expected for a male with a TBM of 70 kg and height of 1.76 m [26]. Theory-based allometric scaling can be used to compare CL values in terms of a standardised NFM value, most widely expressed for a 70 kg individual, with an allometric exponent of ¾. This is shown in Eq. (5). The effects of size and body composition on drug pharmacokinetics can be predicted using NFM, allometric theory and the separation of body mass into its fat and fat-free components [27].
2.4 Random Effects
2.4.1 Individual Parameter Model
Population parameter variability (PPV) was accounted for using an exponential model for the random effect variables (η) (Eq. 6). This assumes a log-normal distribution and avoids parameter estimates falling below biologically plausible values. Each of these random variables (η) was assumed to have mean of 0 and a variance denoted by ω2, which was estimated. Between-subject parameter variability is expressed as an apparent coefficient of variation [CV (%)] obtained from the square root of the variance estimate.
where Pij is the parameter for the ith individual on the jth occasion, and Ppop is the population parameter estimate for the parameter P (e.g. CL, V).
The covariance between two elements of η (e.g. CL and V) is a measure of the statistical association between these two variables. Their covariance is related to their correlation (R):
2.4.2 Observational Model
Residual unidentified variability (RUV) was modelled using both proportional (RUVPROP) and additive residual (RUVADD) errors. The between-subject variability (ηRUV,i) of RUV components was also estimated:
All parameters were estimated using the first-order conditional interaction estimate method of NONMEM 7.5. The convergence criterion was three significant digits.
2.5 Model Selection
The minimum value of the objective function (OBJ [− 2log-likelihood (− 2LL)] provided by NONMEM served as a guide during model building. Model selection was also based on parameter plausibility and prediction-corrected visual predictive check (pcVPC) plots [28]. For two nested models, a decrease in the minimum value of the objective function (ΔOBJ) of 3.84 points for an added parameter was considered significant at the 0.05 level. One hundred nonparametric bootstrap replications were used to estimate parameter means and 95% confidence intervals (CIs) as a measure of parameter uncertainty [29]. Results from the population models are presented as parameter estimates, together with their 95% CIs.
2.6 Quality of Fit
The quality of fit during model building was assessed by visual inspection of diagnostic plots (e.g. observed versus predicted concentrations and residual plots). Bootstrap methods, incorporated within the Wings for NONMEM program, provided a means to evaluate parameter uncertainty [30]. A total of 100 replications were used to estimate parameter confidence intervals. A pcVPC [31], a modelling tool that estimates the concentration prediction intervals and graphically superimposes these intervals on observed concentrations after a standardised dose, was used to evaluate how well the model predicted the distributions of the observed acetaminophen and ibuprofen concentrations. The pcVPC accounts for differences in covariates such as dose, weight, height and sex within the study population [32]. Observations and simulations are multiplied by the population baseline value divided by the individual-estimated baseline.
In any model, the quality of the individual parameter estimate will depend heavily on the observed data available. For example, sparse data can result in reduced variance (ω2) of parameter estimates and distortions of the distribution shape. If no data are available on a particular individual, the individual’s estimate will be equal to the population value; the variance shrinks towards zero as the quantity of information at the individual level diminishes—a phenomenon defined as η shrinkage (Shη). The shrinkage (Sh\(\eta\)%) was calculated using
where SD approximates the standard deviation. When there is no shrinkage, the model is correct, and individual data are sufficiently abundant for individual parameter estimation. Data contain virtually no information about these parameters when shrinkage is 100% and the individual parameter values approach the typical parameter value.
2.7 Simulation
Demographic information from Studies 1 to 4 were pooled and resampled with replacement 1000 times to create a simulation datafile. Acetaminophen and ibuprofen concentrations were simulated at 5, 15, 30 and 45 min and 1, 1.25, 1.5, 2, 3, 6, 8, 10 and 12 h after drug administration. The final pharmacokinetic models for acetaminophen and ibuprofen were used to simulate the maximal concentration (CMAX) and the time taken to reach maximal concentration (TMAX) after acetaminophen 1000 mg and ibuprofen 300 mg for tablet, suspension and sachet formulations.
The impact of absorption differences attributable to formulation and the effects on plasma concentration and visual analogue pain score (0–10) were assessed using simulation. A pharmacodynamic model for combined acetaminophen and ibuprofen (Supplementary Table S4) [33] was used to simulate the concentration–response relationship for each drug formulation in the fasting states. Drug effect was measured as a reduction in a 10-point pain scale. A reduction of the pain score by 2 points was considered clinically relevant [34].
Pharmacokinetic parameter estimates were obtained from the current final acetaminophen and ibuprofen models. Simulations were performed in Berkeley Madonna (Robert Macey and George Oster of the University of California, Berkeley, USA) for a typical 70 kg individual given acetaminophen 1000 mg or ibuprofen 300 mg.
3 Results
There were with 6,095 acetaminophen and 6,046 ibuprofen concentrations amenable for modelling from 116 individuals. Demographic details for the pooled study participants are shown in Table 1. The distributions of participant age and weight are shown in Supplementary Fig. S1. A two-compartment disposition model better described time–concentration data for acetaminophen and ibuprofen than a one-compartment model (ΔOBJ − 3309.885 and − 624.355, respectively, for two additional parameters). The model-building steps and the influence of each covariate on OBJ are shown in Supplementary Table S1.
Allometric scaling of NFM was the best covariate on acetaminophen clearance, with a factor for fat (FFAT) on CL of 0.816. Acetaminophen volume of distribution was best described using TBW. Allometric scaling of NFM was the most suitable covariate for ibuprofen clearance and volume of distribution. Ibuprofen FFATs on CL and Vd were 0.863 and 0.718, respectively.
The final pharmacokinetic parameter estimates are shown in Table 2 for acetaminophen and Table 3 for ibuprofen. The correlation between pharmacokinetic subject variability for clearance (CL), volume of distribution of the central compartment (V1), intercompartmental clearance (Q2) and volume of distribution of the peripheral compartment (V2) is shown in Supplementary Table S2. Prediction-corrected visual predictive checks for the final acetaminophen and ibuprofen models are shown in Fig. 1. Further diagnostic plots are available in the Supplementary Material.

Prediction-corrected visual predictive check (pcVPC) for the acetaminophen and ibuprofen pharmacokinetic models. Models were developed using pooled acetaminophen and ibuprofen plasma concentrations. Plots show median (solid lines) and 90% intervals (dashed lines). The left plots show all prediction-corrected observed acetaminophen and ibuprofen concentrations. Right plots show prediction-corrected percentiles (10%, 50% and 90%) for observations (grey dashed lines) and predictions (red dashed lines) with 95% confidence intervals for prediction percentiles (median, pink shading; 5th and 95th percentiles, blue shading)
Addition of a lag time on oral administration improved the base model for acetaminophen and ibuprofen (ΔOBJ: − 1750.81 and − 636.468, respectively). The final model quantified the influence of feeding on the absorption parameters TLAG and T1/2 ABS for each drug formulation. When fed, acetaminophen T1/2 ABS for the tablet formulation was 1.9-fold slower than the tablet under fed conditions, and the lag time on the tablet formulation increased 4.6-fold under fed conditions. The increase in lag time attributed to feeding was not as pronounced with the sachet or suspension formulation (2-fold and 3-fold, respectively). Tablet ibuprofen administered in fed states resulted in a slower (1.6-fold) T1/2 ABS compared with that observed with a tablet administered in the fasted state. Feeding increased T1/2 ABS 2.5-fold and 4-fold with suspension and sachet formulations. The largest increase in lag time with feeding was 3.7-fold, and was associated with the ibuprofen tablet formulation. Bioavailability on oral formulations was 86% for acetaminophen and 94% for ibuprofen.
Simulated CMAX and TMAX for acetaminophen and ibuprofen oral formulations in the fed and fasting states are shown in Table 4. Acetaminophen CMAX is achieved more rapidly under fasting conditions for all oral formulations (e.g. tablet TMAX 0.61 h versus 0.88 when fed). Ibuprofen tablet TMAX is more rapid under fasting conditions than under fed conditions (1.2 h versus 0.94 h). Acetaminophen and ibuprofen suspension and sachet simulated TMAX values are shorter in fasting states compared with fed states.
Simulation was used to describe the pain time course after drug administration (Supplementary Fig. S2). These demonstrate that although CMAX may be greater and TMAX shorter for the sachet than those predicted for the tablet, effect (pain relief score) is similar for both formulations. Time to peak effect for sachet is shorter than for the tablet formulation (e.g. 10 min for paracetamol in the fasted state). The time taken to achieve a meaningful reduction in pain score (e.g. 2 points on a visual analogue pain score of 0–10) differed by only 5–10 min across all formulations for acetaminophen and ibuprofen (Supplementary Table S4).
4 Discussion
Population pharmacokinetic parameters were estimated for acetaminophen and ibuprofen using two-compartment disposition models after pooling time–concentration data obtained from healthy fed and fasted adults. The influence of feeding on absorption was described using a pharmacokinetic compartmental method. Data were available following intravenous and oral routes of administration, enabling the estimation of the absorption parameters bioavailability, absorption half-life and lag time (F, T1/2 ABS and TLAG, respectively). Oral routes included tablet, sachet and oral suspension formulations containing both acetaminophen and ibuprofen, allowing quantification of the influence of feeding on absorption parameters for each oral formulation. Clearance and volume of distribution are the primary pharmacokinetic parameters used to determine maintenance and loading dose. Allometric scaling of total body weight [25] is an important covariate used to describe the variability associated with these pharmacokinetic parameters between individuals [21].
Clearances and volumes of distribution are independent of dose, formulation and feeding. Studies investigating drug absorption frequently express clearances confounded by bioavailability (e.g. CL/F) or fail to scale parameters to a typical individual (e.g. 70 kg), which creates confusion when comparing values between studies [35]. Acetaminophen and ibuprofen pharmacokinetics are extensively described in the literature for children and adults [1, 36,37,38,39]. The clearances and volumes of distribution we report are similar to those in other studies of acetaminophen and ibuprofen in adults [37]. The bioavailability for acetaminophen in this analysis was 87%, and that for ibuprofen was 93%. Previous noncompartmental analyses of these data reported similar relative bioavailabilities: 93.8% for acetaminophen and 96.4% for ibuprofen [36].
The acetaminophen absorption half-life was 11.5 min. This increased approximately twofold for all oral formulations in fed states; the most pronounced effect was observed with the suspension formulation. Ibuprofen yielded similar results. The absorption half-life was 27 min, which increased two- to fourfold for all oral formulations under fed states. These estimates align with a study in postoperative adults: the absorption half-lives for acetaminophen were 9.6 min and 51 min for ibuprofen when administered in combination [1]. Paracetamol is a Biopharmaceutical Classification System (BCS) Class 1 drug with high permeability and solubility, while ibuprofen is a BCS class II drug [40], meaning it is poorly soluble but highly permeable. This may be reflected in the absorption half-life of ibuprofen, which is twice that of acetaminophen.
The absorption delay (lag time) for oral acetaminophen and ibuprofen formulations was protracted in fed states. The largest delay in absorption was associated with tablet formulations for acetaminophen and ibuprofen (4.60-fold and 3.65-fold, respectively). Decreased or delayed drug absorption due to changes in gastric emptying can be attributed to factors other than feeding. When administered concurrently with paracetamol, narcotic analgesics (diamorphine, pethidine) delayed the gastric emptying rate, thereby decreasing paracetamol absorption [41]. Absorption of nasogastric elixir paracetamol in adults undergoing cardiac surgery was delayed compared with healthy adults [42]. The absorption of oral oxycodone is also delayed after cardiac surgery, with the most profound delay (when compared with preoperative administration) observed on the first postoperative day [43].
CMAX and TMAX are noncompartmental parameters used to describe drug pharmacokinetics. These noncompartmental parameters CMAX and TMAX demonstrate similar trends of delayed absorption due to feeding, which can be quantified by increases in T1/2 ABS and TLAG. Simulated CMAX values after acetaminophen 1000 mg and ibuprofen 300 mg align with reported values [37, 44,45,46,47]. Published estimates of TMAX for both drugs are often associated with large variability. This may be due to limitations in study size and the frequency of samples obtained during the absorption phase. Simulation overcomes this problem: plasma concentrations can be predicted at frequent time points in a large number of hypothetical individuals, and CMAX and TMAX can be calculated from dose, bioavailability, clearance and volume of distribution. Formulation-related differences in T1/2 ABS and TLAG affect CMAX and TMAX.
The impact of absorption differences between formulations on simulated pain scores was minimal. Concentration is the factor that drives drug effect. Acetaminophen sachet and suspension formulations were associated with a 5 min shorter time to achieve a pain score reduction of 2 points (VAS 0–10) compared to the tablet. However, the peak effect was similar for all formulations (Supplementary Fig. S2). The variability associated with both pharmacokinetic and pharmacodynamic parameter estimates implies that these small population differences in observed onset of analgesia or peak effect will have little impact on an individual patient [48].
Pooling acetaminophen and ibuprofen data across these studies where both intravenous and oral time–concentration data were available allowed the estimation of the absorption parameters F, T1/2 ABS and TLAG. The precision associated with ibuprofen bioavailability and lag time is poor. This may be attributed to inadequate sampling during the early absorption phase. This study was conducted in typical healthy individuals. These changes in absorption parameters we observe may be altered in different cohorts such as the elderly, obese or those with impaired hepatic function.
We demonstrate the utility of compartmental models to quantify the influence of feeding on the commonly used analgesics acetaminophen and ibuprofen. Modelling and simulation can augment traditional metrics (e.g. CMAX and TMAX) used to describe acetaminophen and ibuprofen pharmacokinetics.
Fat mass, a covariate, had an influence on acetaminophen clearance estimation and on both clearance and volume of distribution for ibuprofen. However, obesity is also associated with concomitant pathology, which can affect pharmacokinetic parameter estimates [49], and further study of an obese population is required to substantiate the dosing prediction for that population.
5 Conclusion
Fat mass is an important covariate to describe acetaminophen and ibuprofen pharmacokinetics. The absorption half-lives of acetaminophen and ibuprofen were increased in fed states. The delay in absorption, quantified by a lag time, was protracted for both drugs in fed individuals. There were minimal predicted differences in effect for both acetaminophen and ibuprofen across all formulations in fed states.
References
Merry AF, Gibbs RD, Edwards J, Ting GS, Frampton C, Davies E, Anderson BJ. Combined acetaminophen and ibuprofen for pain relief after oral surgery in adults: a randomized controlled trial. Br J Anaesth. 2010;104:80–8.
Hannam JA, Anderson BJ, Potts A. Acetaminophen, ibuprofen, and tramadol analgesic interactions after adenotonsillectomy. Pediatr Anesth. 2018;28:841–51.
Anderson B. Paracetamol (acetaminophen): mechanisms of action. Pediatr Anesth. 2008;18:915–21.
Aronoff DM, Oates JA, Boutaud O. New insights into the mechanism of action of acetaminophen: its clinical pharmacologic characteristics reflect its inhibition of the two prostaglandin H2 synthases. Clin Pharmacol Ther. 2006;79:9–19.
Burian M, Geisslinger G. COX-dependent mechanisms involved in the antinociceptive action of NSAIDs at central and peripheral sites. Pharmacol Ther. 2005;107:139–54.
Wick EC, Grant MC, Wu CL. Postoperative multimodal analgesia pain management with nonopioid analgesics and techniques: a review. JAMA Surg. 2017;152:691–7.
Sharma CV, Mehta V. Paracetamol: mechanisms and updates. Contin Educ Anaesthesia Crit Care Pain. 2013;14:153–8.
Rainsford KD. Ibuprofen: pharmacology, efficacy and safety. Inflammopharmacology. 2009;17:275–342.
Hannam J, Anderson BJ. Explaining the acetaminophen-ibuprofen analgesic interaction using a response surface model. Paediatr Anaesth. 2011;21:1234–40.
Daniels SE, Atkinson HC, Stanescu I, Frampton C. Analgesic efficacy of an acetaminophen/ibuprofen fixed-dose combination in moderate to severe postoperative dental pain: a randomized, double-blind, parallel-group, placebo-controlled trial. Clin Therapeut. 2018;40:1765-1776.e1765.
Ong CK, Seymour RA, Lirk P, Merry AF. Combining paracetamol (acetaminophen) with nonsteroidal antiinflammatory drugs: a qualitative systematic review of analgesic efficacy for acute postoperative pain. Anesth Analg. 2010;110:1170–9.
Moore RA, Derry S, Aldington D, Wiffen PJ. Single dose oral analgesics for acute postoperative pain in adults—an overview of Cochrane reviews. Cochrane Database Syst Rev. 2015;2015:CD008659.
Toothaker RD, Welling PG. The effect of food on drug bioavailability. Annu Rev Pharmacol Toxicol. 1980;20:173–99.
Eleveld DJ, Colin P, Absalom AR, Struys M. Pharmacokinetic-pharmacodynamic model for propofol for broad application in anaesthesia and sedation. Br J Anaesth. 2018;120:942–59.
Eleveld DJ, Proost JH, Absalom AR, Struys MM. Obesity and allometric scaling of pharmacokinetics. Clin Pharmacokinet. 2011;50:751–3.
Eleveld DJ, Proost JH, Vereecke H, Absalom AR, Olofsen E, Vuyk J, Struys M. An allometric model of remifentanil pharmacokinetics and pharmacodynamics. Anesthesiology. 2017;126:1005–18.
Anderson BJ, Holford NH. Getting the dose right for obese children. Arch Dis Child. 2017;102:54–5.
Anderson BJ, Holford NH. What is the best size predictor for dose in the obese child? Paediatr Anaesth. 2017;27:1176–84.
Holford NHG, Anderson BJ. Allometric size: the scientific theory and extension to normal fat mass. Eur J Pharm Sci. 2017;109:S59–64.
Janmahasatian S, Duffull SB, Ash S, Ward LC, Byrne NM, Green B. Quantification of lean bodyweight. Clin Pharmacokinet. 2005;44:1051–65.
Allegaert K, Olkkola KT, Owens KH, Van de Velde M, de Maat MM, Anderson BJ, The PACIA Study Group. Covariates of intravenous paracetamol pharmacokinetics in adults. BMC Anesthesiology. 2014;14:77.
Sheiner LB. The population approach to pharmacokinetic data analysis: rationale and standard data analysis methods. Drug Metab Rev. 1984;15:153–71.
Holford NHG. A size standard for pharmacokinetics. Clin Pharmacokinet. 1996;30:329–32.
Anderson BJ, Meakin GH. Scaling for size: some implications for paediatric anaesthesia dosing. Paediatr Anaesth. 2002;12:205–19.
Anderson BJ, Holford NH. Mechanism-based concepts of size and maturity in pharmacokinetics. Annu Rev Pharmacol Toxicol. 2008;48:303–32.
Holford NHG, Anderson BJ. Allometric size: the scientific theory and extension to normal fat mass. Eur J Pharm Sci. 2017;109S:S59–64.
Anderson BJ, Holford NHG. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab Pharmacokinet. 2009;24:25–36.
Nguyen TH, Mouksassi MS, Holford N, Al-Huniti N, Freedman I, Hooker AC, John J, Karlsson MO, Mould DR, Perez Ruixo JJ, et al. Model evaluation of continuous data pharmacometric models: metrics and graphics. CPT Pharmacometr Syst Pharmacol. 2017;6:87–109.
Parke J, Holford NH, Charles BG. A procedure for generating bootstrap samples for the validation of nonlinear mixed-effects population models. Comput Methods Progr Biomed. 1999;59:19–29.
Efron B, Tibshirani R. Bootstrap methods for standard errors, confidence intervals, and other measures of statistical accuracy. Stat Sci. 1986;1:54–77.
Post TM, Freijer JI, Ploeger BA, Danhof M. Extensions to the visual predictive check to facilitate model performance evaluation. J Pharmacokinet Pharmacodyn. 2008;35:185–202.
Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. J AAPS. 2011;13:143–51.
Hannam JA, Anderson BJ, Potts A. Acetaminophen, ibuprofen, and tramadol analgesic interactions after adenotonsillectomy. Paediatr Anaesth. 2018;28:841–51.
Bahreini M, Safaie A, Mirfazaelian H, Jalili M. How much change in pain score does really matter to patients? Am J Emerg Med. 2020;38:1641–6.
Holford NH, Anderson BJ. Why standards are useful for predicting doses. Br J Clin Pharmacol. 2017;83:685–7.
Atkinson HC, Stanescu I, Frampton C, Salem II, Beasley CPH, Robson R. Pharmacokinetics and bioavailability of a fixed-dose combination of ibuprofen and paracetamol after intravenous and oral administration. Clin Drug Investig. 2015;35:625–32.
Forrest JAH, Clements JA, Prescott LF. Clinical pharmacokinetics of paracetamol. Clin Pharmacokinet. 1982;7:93–107.
Anderson BJ, Woollard GA, Holford NHG. A model for size and age changes in the pharmacokinetics of paracetamol in neonates, infants and children. Br J Clin Pharmacol. 2000;50:125–34.
Davies NM. Clinical pharmacokinetics of ibuprofen. Clin Pharmacokinet. 1998;34:101–54.
Álvarez C, Núñez I, Torrado JJ, Gordon J, Potthast H, García-Arieta A. Investigation on the possibility of biowaivers for ibuprofen. J Pharm Sci. 2011;100:2343–9.
Nimmo WS, Wilson J, Prescott LF. Narcotic analgesics and delayed gastric emptying during labour. The Lancet. 1975;305:890–3.
Schuitmaker M, Anderson BJ, Holford NHG, Woollard GA. Pharmacokinetics of paracetamol in adults after cardiac surgery. Anaesth Intensive Care. 1999;27:615–22.
Valtola A, Morse JD, Florkiewicz P, Hautajärvi H, Lahtinen P, Musialowicz T, Anderson BJ, Ranta V-P, Kokki H. Bioavailability of oxycodone by mouth in coronary artery bypass surgery patients—a randomized trial. J Drug Assess. 2020;9:117–28.
Langford RA, Hogg M, Bjorksten AR, Williams DL, Leslie K, Jamsen K, Kirkpatrick C. Comparative plasma and cerebrospinal fluid pharmacokinetics of paracetamol after intravenous and oral administration. Anesthesia Analgesia. 2016;123:610–5.
Nielsen JC, Bjerring P, Arendt-Nielsen L. A comparison of the hypoalgesic effect of paracetamol in slow-release and plain tablets on laser-induced pain. Br J Clin Pharmacol. 1991;31:267–70.
Kale P. Pharmacokinetics and bioavailability of single dose ibuprofen and pseudoephedrine alone or in combination: a randomized three-period, cross-over trial in healthy Indian volunteers. Front Pharmacol. 2014;5:98–98.
Shin D, Lee SJ, Ha Y-M, Choi Y-S, Kim J-W, Park S-R, Park MK. Pharmacokinetic and pharmacodynamic evaluation according to absorption differences in three formulations of ibuprofen. Drug Des Devel Ther. 2017;11:135–41.
Holford NH, Buclin T. Safe and effective variability—a criterion for dose individualization. Ther Drug Monit. 2012;34:565–8.
Cortinez LI, Anderson BJ, Holford NH, Puga V, de la Fuente N, Auad H, Solari S, Allende FA, Ibacache M. Dexmedetomidine pharmacokinetics in the obese. Eur J Clin Pharmacol. 2015;71:1501–8.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
JDM and BJA have no conflicts of interest to declare. HA and IS are employees and shareholders in AFT Pharmaceuticals.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. This work was funded by AFT Pharmaceuticals Limited, Auckland, New Zealand. This work was performed using a license for NONMEM granted by ICON to the Australian Centre of Pharmacometrics. The Australian Centre for Pharmacometrics is an initiative of the Australian Government as part of the National Collaborative Research Infrastructure Strategy. James Morse is supported in part by the New Zealand Society of Anaesthetists Ritchie Prize.
Ethics approval
These studies were approved by the Jordan Food and Drug Administration and were registered with the Australian New Zealand Clinical Trial Registry (AFT-MXIV-01—ACTRN12614000809639; AFT-MXIV-06—ACTRN12615001208594; AFT-MX-14a—ACTRN12616000418471; AFT-MX-14b—ACTRN12616000419460).
Consent to participate
Written informed consent was given by all participants.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Consent for publication
Not applicable.
Code availability
NM-TRAN control streams are available on request.
Author contributions
All authors whose names appear on the submission made substantial contributions to the conception or design of the work, the acquisition, analysis, interpretation of data used in the work. All authors revised it critically for important intellectual content, approved the version to be published; and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Morse, J.D., Stanescu, I., Atkinson, H.C. et al. Population Pharmacokinetic Modelling of Acetaminophen and Ibuprofen: the Influence of Body Composition, Formulation and Feeding in Healthy Adult Volunteers. Eur J Drug Metab Pharmacokinet 47, 497–507 (2022). https://doi.org/10.1007/s13318-022-00766-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-022-00766-9