
Summary
Clinical trials are subject to an ever-evolving landscape of treatment options, regulatory requirements, socioeconomic conditions, and paradigms of scientific research. In this opinion paper, we illustrate current methods and topics with a focus on clinical trial designs, conduct and modes of collaboration. The example of successful clinical breast cancer research in Austria demonstrates the opportunities, but also the challenges for academic research. We also investigate potential pitfalls, and suggest new ideas and strategies on how to increase practicability along with transparency and fairness, but ultimately also scientific and clinical relevance of clinical trials in oncology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Clinical trials have become the mainstay of knowledge generation in modern applied medical sciences and are an essential cornerstone in continuously advancing patient care. In this article, we analyze some of the strengths and weaknesses of current methodology and governance. We also provide reflection and some suggestions for the future development of this important part of constantly redefining and improving the so-called standard of care. In this up-to-date summary and opinion paper, we investigate how potential improvements can be integrated into contemporary practice of clinical trial design, conduct and reporting. In order to provide impulses and “food-for-thought” to readers and clinical trial enthusiasts, we aim to provide a wide range of aspects that in our opinion require consideration when conducting clinical research—this however leads to some inevitable superficiality; a real in-depth analysis of all topics mentioned would result in a book format and goes beyond the scope of this article.
Based on the practical perspectives and experience of decades of internationally successful clinical and translational breast cancer research, we discuss the medical/clinical as well as operational view of clinical trials. We argue that the most important strategic goal of clinical research should always be to ensure that patient-relevant issues are studied. Ultimately, innovation must be brought to patients in an effective and timely manner. How this can be achieved is depicted in this article in various topics and proposed contemporary methods, supplemented by specific examples. Further emphasis is placed on the building of professional operational structures and development of time- and cost-effective strategies to conduct academic clinical research—and thus to remain capable to (both scientifically and operationally) compete with industry-sponsored trials. Focusing on the most pressing scientific questions, focusing on key data collection, and omitting an over-burden on involved research teams are over-arching aims if we want to adapt clinical trials to a fast-paced regulatory environment, and limitations in study funding for academia.
Patient-centric approach and less restrictive trial designs
Clinical research would not be possible without volunteers who donate their valuable time, data and biospecimen as well as their trust that clinical trials are conducted at a high-quality scientific and operational level. Recently, an increased focus has been placed on the involvement of participants in the development and conduct of clinical trial designs. A patient-centric approach to clinical research may positively influence study recruitment as well as decrease the loss of participants during the study and prolonged study follow-up and subsequently enable the research teams to better tackle the important topic of patient retention [1]. As it is crucial to minimize the burden on patients during participation in clinical trials, the incorporation of patient advocacy feedback when developing a clinical trial protocol seems worthwhile. These measures might include patient representatives being able to provide input for planned assessments and study schedules but also being nominated to become members of steering committees throughout the life cycle of a clinical study to support sponsors and caregivers in the essential task of retaining participants on the project.
Based on the advancement of our understanding of diseases, there is currently a trend towards increased complexity of trial designs and—in cancer research towards rare tumor mutations and special patient populations, which are difficult to apply in routine care—a paradigm shift towards less restrictive trial designs and eligibility criteria, and a subsequent reduction of inclusion and exclusion criteria to a minimum seems to be advisable. Developing trial designs that are more inclusive in real-world populations among broader populations might further increase the practical application of clinical research results, while significantly increasing patient recruitment [2, 3].
At the Austrian Breast and Colorectal Cancer Study Group (ABCSG), we closely collaborate with Austrian and European patient advocacy groups (e.g., www.europadonna.org), by inviting patient representatives to have formal votes on study-specific steering committees and to provide them a platform to share the patient’s view, for example in the course of annual group meetings. It is furthermore intended to mimic real-world populations and not just create rather specific interventions and cohorts by conducting noninterventional studies and registries in breast cancer, pancreatic cancer, and colorectal cancer.
Decentralization and regional/institutional diversity for better patient engagement
The commitment of compliant trial participants might be further increased by using decentralized trial approaches [4] that can be better integrated into daily routines, such as telemedicine visits and remote data collection via wearable technologies (e.g., smart phones, smart watches, tablets) [5]. A decentralized set-up facilitates data collection from geographically large and diverse populations and may therefore have a positive effect on trial recruitment as well as ethnical diversity. As clinical trials are mainly conducted in academic medical centers, and respective specialized facilities may not be available in all remote areas, decentralization should be helpful to include patients in clinical trials living in rural areas.
Experience from large breast cancer studies at ABCSG in the last 30 years [6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22] showed that smaller, rural clinics significantly contributed to study enrollment, and that less bureaucratic administrative burden associated with them—compared to larger university/research hospitals—often provided for a rather quick set-up, and highly successful track record of enrollment and compliance. In addition, collaborations and scientific exchange with smaller medical practices that can refer patients to clinical research facilities or clinical research projects are an important step leading to broader patient inclusion [23, 24].
In addition to outreach measures to the geographical periphery and usually less integrated clinics and healthcare providers in countryside regions, it is essential to generally balance and represent as many demographic subgroups in clinical research: a lack of “subject diversity” in clinical trials and overly “homogenous” trial participants (e.g., only one gender, race/ethnicity, age group) may result in a nongeneralizable—and thus incomplete—resulting dataset. On the other hand, inclusion of a diverse set of individuals may lead to more robust and complete data, while broadening the understanding of, for example, ethnic differences in treatment response. A fair balance in ethnicity is an increasingly important topic in enrollment for multiethnic, large-scale societies such as the USA, with communities that consist of heterogenous backgrounds, both in terms of genetic predispositions and variance as well as socioeconomic possibilities [25]. While we might have fewer African–European citizens in European countries compared to the US, our ethnical diversity is also increasing based on recent migration and refugee events—these new “subpopulations” deserve particular attention since they are especially endangered to be underrepresented in clinical trials, for a variety of reasons, including limited access to care, cultural differences, and many others [26].
In general, language barriers, mistrust in clinical research, but certainly also lack of information (or: misinformation in regard to clinical research) as well as time and resource constraints may negatively influence patients to participate in clinical research studies. Consequently, increasing the awareness of research studies, also among minorities and low-income communities, is pivotal as well as taking time when discussing the possible participation in a study with a patient and provide credible, clear, and trustworthy information. For example, provision of informed consent forms in the main spoken languages of the country, prepared in a comprehensible lay language manner, in combination with diverse healthcare professionals with different ethnical backgrounds as well as collaborations with smaller care units to win the trust of marginalized groups, may be helpful strategies.
Involvement and education of diverse healthcare teams
Diversity and inclusion efforts within professional healthcare teams—not only in the potential trial population—might further increase patient recruitment and potentially study compliance in (minority) populations, as healthcare studies show that patients generally fare better when care was provided by more diverse teams [27]. In addition, there is evidence suggesting that more diverse teams lead to improvements in innovations, team communications, and improved risk assessment. In terms of inclusion of various healthcare teams, ABCSG—in addition to planning, conducting, and analyzing clinical research studies—strives for the second main aim of distributing trial results and knowledge among the scientific (i.e., academic treatment centers) but also a broader medical community within community based or country-side hospitals. In order to achieve this, trial results and the current state-of-the-art are not only communicated via high-level scientific publications and presentations but also using a wide array of continuing education events. In these events, there is an active effort to keep a fair balance considering the expertise, age, and gender among speakers and expert panelists; in addition, the surrounding communication efforts aim to attract an audience of healthcare professionals from various educational levels, backgrounds, and age groups, also offering tailored events for more rural areas and smaller hospitals as well as the “next generation” with the involvement of a dedicated task force led by young, female investigators.
Digitalization and artificial intelligence
In this context, digitization plays a major role in providing a more inclusive and accessible clinical trial landscape, while reducing the workload of healthcare professionals through automatizing time-consuming processes [4]. Digitized tools, such as the use of electronic medical records (EMRs), are already in use for retrospective data collection and introduced in retrospective, noninterventional study designs. EMRs can further be used in the screening process, i.e., to check for eligible patients according to the information available in the database and to subsequently perform a remote contact and consenting of potential participants. In general, online informed consent procedures (via video calls, etc.) are still facing legal and regulatory challenges in some environments (e.g., keeping in mind rather strict General Data Protection Regulation (GDPR) guidelines in the EU; https://eur-lex.europa.eu/eli/reg/2016/679/2016-05-04) but seem to be a robust new method to include patients in remote areas and maintain patient retention (e.g., reduction of lost to follow-up patients as well as re-consenting possibilities regarding further use of already collected biological samples with no additional on-site visits of patients).
The use of further digitized patient-related tools such as ePROs or eDiaries also become increasingly common in new emergent clinical research trials [28]. Still, challenges remain as digitized approaches might reduce study populations to younger or more technically versed participants, and remote data collection designs require active participation from study subjects (i.e., responsibility of data collection is in the hands of the patient). Importantly, appropriate steps to ensure data protection and obtaining informed consent in accordance with applicable regulations need to be ensured when implementing digitized elements in clinical trial designs.
Another new emerging strategy in the digital realm is the implementation of machine-learning-assisted methods as well as use of artificial intelligence (AI). Major advantages of integrating these tools include the automation of time-consuming, repetitive tasks alongside the reduction of human error. Practical applications in cancer clinical research are manifold and include, among others, use in diagnostics [29] and screening, onco-imaging (using imaging assessment data), pathology (using digitized pathological slides) for diagnosis, grading, prognostication, and the use in translational oncology to further study molecular pathways with the aim to develop novel therapeutic interventions.
Despite the wide range of possibilities that come with the incorporation of AI in clinical research and medical care, several challenges remain in regard to the natural and unique heterogeneity of the human body between individuals, but are also present in behavior and institutions as well as factors related to the technology and underlying algorithms of AI itself. Therefore, precise clinical research studies and study protocols released by ethics committees need to be further developed and conducted that eventually prove the actual benefit use of the emerging opportunities, while maintaining a sound balance of patient rights and data integrity. In current and planned collaborative projects with biotech and AI companies, ABCSG explores projects that include machine learning endpoints, for example, using digitized pathology slides by using a machine-learning-assisted development of breast cancer signatures and patterns from these images that can increase prognostic and potentially predictive value of such automated imaging tests. As for AI-based language processing tools such as ChatGPT (OpenAI, San Francisco, CA, USA)), there is currently only limited use in the context of clinical and development of clinical trial design due to the confidential and sensible nature of the associated data. The natural tension between opportunities and chances of using AI for clinical research and observing basic data protection rules in order to guarantee the necessary privacy but also sensitive health-relative information. Some suggestions to define this corridor have recently been proposed [30].
Translational research and biomarker-guided therapy
Translational research is an important topic in the context of current clinical research and a cornerstone of ABCSG’s oncology portfolio of scientific projects. The inclusion of translational research questions and biosample collection in the early stage of the development of clinical trial protocol development is of utmost importance. During that protocol development stage, key translational research objectives should be defined in order to collect and process biological samples correctly and use appropriately for future projects. Genomic assays that provide treatment guidance based on the genomic composition of each patient as well as individual signatures in other -omics fields (e.g., proteomics) support a tailored approach for medical care. With costs for RNA and DNA sequencing continuing to decrease, there is the opportunity to make individualized diagnosis and treatment options more accessible (first in clinical research studies and after testing and validation in these projects, companies will have an incentive to provide such tests on a larger and more affordable scale outside of clinical studies).
Biomarker-guided participant screening and inclusion, as well as treatment decisions, are increasingly integrated into prospective trial designs, with less-invasive biopsy options such as circulating tumor DNA (ctDNA), or tumor-infiltrating lymphocytes (TILs) being assessed in different entities and subtypes of cancer for their practical application in clinical care.
As a result of the “abcsg.research” initiative, we have shown on multiple instances how close collaboration with companies in biotech and translational research led to the development and validation of tests by making good use of ABCSG’s invaluable collection of biospecimen as well as long-term follow-up and survival data of (neo)adjuvant breast cancer patient cohorts [31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47].
Moving forward, new study designs should already integrate the donation of biosamples (tumor tissue and blood samples) to allow not only for new concepts such as ctDNA-guided therapy, but also for the use in future research projects that may address questions with quickly emerging technologies later on.
Trends in clinical statistics and trial designs
Among the variety of developments within the broad field of biostatistics, we would like to mention a few topics which attracted high attention in recent years. With the publication of the estimand framework [48], a huge step forward has been made in understanding and quantifying treatment effects. Most importantly, the framework recognizes that the different handling of so-called “intercurrent events” (i.e., events that occur after baseline but prior to the endpoint of interest) can alter the scientific question that is addressed. In other words, two trials with the exact same endpoint definition could address different objectives when both do not handle intercurrent events in the same way (e.g., ignoring vs censoring for early drug discontinuations [49]). Since then, this concept has found wide recognition in the statistical community (see, for example, [50,51,52,53]) and is on its way to be routinely used in study protocols and analysis plans to eventually help in the definition of the scientific questions and understanding of clinical data.
Furthermore, as patient involvement into trial designs becomes more important, it might also be time to move away from the traditional way of assessing the benefit vs harm of a drug to a more patient-centric assessment. In the past and still today, efficacy and safety endpoints are analyzed separately, which is mainly because of the limitations of traditional statistical methods. But this has been criticized in the past and a more “patient-centric” approach has been suggested where the efficacy and toxicity of a drug is combined into a single, quantitative measure [54]. With the invention of a “new” statistical method, called “generalized pairwise comparisons”, the groundwork has been laid for enabling the combined analysis of efficacy and safety and many extensions have been made recently [55,56,57]. And importantly, the effect can then be summarized with the so-called net treatment benefit (NTB). The NTB is an absolute treatment effect measure which directly addresses the benefit vs harm issue, and it has a number of advantages. To name only a few: it does not rely on the proportional hazards assumption, it is probably more meaningful than the classic hazard ratio as it provides the net chance of a longer survival [58] and it overcomes limitations of standard time-to-first-event analysis (i.e., it prioritizes more relevant components of a composite endpoint [59], e.g., a local recurrence is less problematic than a distant recurrence in breast cancer). It is therefore not surprising that this new method has already been suggested for assessing treatment benefit in immuno-oncology [60] and has been used successfully to gain a deeper understanding of the drug effects of FOLFIRINOX and nab-paclitaxel in metastatic pancreatic adenocarcinoma [61, 62].
Furthermore, there is still and active and controversial discussion about optimal primary endpoints in clinical trials. For example, in a very recent commentary, Leary et al. argued that overall survival (OS) and quality of life (QOL) should be the main endpoints [63]. It is difficult to imagine that OS will replace progression-free-survival (PFS) in metastatic clinical trials any time soon (and even less realistic that OS might replace disease-free-survival (DFS) in all its variations) [64, 65], but maybe generalized pairwise comparisons could be useful here in order to bridge the gap between both approaches: with generalized pairwise comparisons OS and PFS can be analyzed simultaneously: death could be the primary event of interest, but a progression event can be the “tie-breaker” between two patients who have not (yet) died, of whom one has already progressed, and the other did not. This would result in a “combined PFS–OS endpoint” analysis, and has the potential to satisfy both needs of the “overall survival” argument. Whether such an innovative statistical approach can serve as viable replacement strategy for traditional trial endpoints needs to be further assessed with simulations and large clinical trial data sets [66].
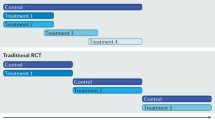
New designs of clinical trials such as platform, umbrella or basket trials have become increasingly popular during recent years. While basket trials aim to assess the same intervention in more than one disease, platform trials seek to evaluate different interventions in one disease and umbrella trials in different subtypes of the same disease [67]. In the field of breast cancer, the I‑SPY series of clinical trials is also another example of a successful co-operation between pharmaceutical companies, academic medical centers, and the US Food and Drug Administration (FDA) as regulatory body: the core trial I‑SPY 2 is a platform trial for neoadjuvant treatment of locally advanced breast cancer; its adaptive-trial design is based on comparisons of new interventions with a standard treatment using Bayesian probability statistics. The great advantage of such adaptive trial designs and multi-arm multistage (MAMS) designs in general is the possibility to react to a quickly changing clinical landscape by replacing an outdated standard of care arm with a more contemporary “new” standard arm. Also, such trial design even allows to close treatment arms (early) that turn out not promising, and rather focus (and invest both resources and the ultimate patient donation of trial participation) on more auspicious treatment arms based on a surrogate endpoint [68, 69].
Regulations and funding of academic research
When discussing the future of clinical trials, we ultimately need to consider current and recently established legal and regulatory frameworks as well. In 2022, the European Clinical Trials Regulation (CTR) came into effect in an effort to harmonize regulatory submissions in a single application instead of requiring applications by each European Union member state individually. The aim is to accelerate the development of medications and bring innovative medicines to broader populations faster. It is yet to be observed, what will be the benefits and pitfalls of the CTR implementation and how it will impact clinical research conduct and whether it will facilitate or place a burden on the conduct of academic clinical research. Based on preliminary experiences in submitting and conducting clinical trials, further adaptions may be warranted to achieve a more harmonized and seamless approval process and avoid burden on smaller, academic research organizations (e.g., those conducting investigator-initiated trials).
On a further note, as the regulatory landscape is changing and becoming more and more complex, time-consuming as well as cost-consuming, it is increasingly challenging to receive sufficient funding for unbiased, independent academic clinical research. Despite some—but painstakingly slow!—recent improvements, Europe still massively lags behind the US in terms of public funding of clinical research (https://www.who.int/observatories/global-observatory-on-health-research-and-development/monitoring/number-of-clinical-trials-by-year-country-who-region-and-income-group) [70,71,72,73]. It is therefore essential (and inevitable!) that close (and transparent!) collaborations between pharmaceutical and/or biotech companies and academic research organizations remain intact, and will even be further strengthened in order to eventually produce the magnitude and variety of unbiased and reliable clinical research data necessary to continuously improve the standard of patient care.
Sharing of such pivotal clinical trial data within the scientific community and with interested researchers who want to address additional questions from the obtained information is another hallmark of successful global collaboration in the future of clinical research. This is supported by many emerging policies and regulations that mandate the public provision of trial results, or even recommend the sharing of (anonymized and aggregated) datasets to stimulate research beyond single sponsors and study groups. At ABCSG, we have successfully conducted “fully academic” projects, albeit in collaboration with pharmaceutical companies, but with ABCSG as legal sponsor [74]. Currently, ABCSG is acting as data custodian in global collaborative studies, with other academic study groups being involved in key elements of clinical trial design and protocol development, even if projects are formally sponsored by pharmaceutical companies (e.g., ABCSG-62/CAMBRIA-2/NCT05952557 is conducted via the transparency model which is described in more detail below).
Building professional operational structures
Large clinical trials for early breast cancer require huge participant numbers, and usually international, often global collaboration. Although sufficient funding for independent clinical research would be the best way to generate unbiased data even of that magnitude, full public or academic funding for such mega-projects is currently unrealistic, thus, collaboration with industry is inevitable. Since the pharmaceutic and biotech industry follows a for-profit model, governance issues of such collaborations need to be discussed and addressed:
In a previous generation of large adjuvant breast cancer trials, international academic collaboration was able to create enough momentum to effectively control governance in many pivotal trials. The Breast International Group (BIG) became a role model for multinational collaboration, and [75] in many successful trials academia effectively “controlled” trial conduct. It is rather undisputed that academic control of clinical trials and their results ensures the avoidance of commercially driven bias, and potential—even somewhat explainable—shareholder value orientation by clinical trial funders [76]. However, some of the challenges for such large collaborations remain the same, irrespective if managed by academia or by for-profit companies: effective governance without overshooting bureaucracy, sufficient balance of timely insight into the factual process of data collection, cleaning and analysis for the noncontrolling collaboration partner and consequent impacts on the trial read out dates.
As a result, it has become less and less popular for industry trial sponsors to allow for sole academic database control, e.g., the most recent generation of large adjuvant trials (investigating the use of Selective Estrogen Receptor Downregulators (SERDs) instead of the previous endocrine standard) has largely regressed into pure industry-controlled setups (e.g., NCT04961996, NCT05514054, NCT05774951). While it is common that industry sponsors outsource total trial conduct—including database control—to global Clinical Research Organizations (CROs), there is a certain level of interdependency between these often also stock exchange listed companies and the pharmaceutic industry as individual trial managements are usually imbedded in large product- or even pipeline-development collaboration models. While there might be—some—academic contribution to scientific and steering committees, and despite there being independent data monitoring committees established in large global trials, eventually the presentation of results, and their interpretation at large, will be confined for most of these multihundred-million investments solely by industry. Of course, we are not suggesting that results be falsified fraudulently, but in itself such “full industry control” constitutes an inevitable conflict of interest, and shows some impact on clinical trial reporting [77].
However, there are successful examples of international academic clinical research: another is the nonprofit academic research organization European Organisation for Research and Treatment of Cancer (EORTC) that also has worked with a variety of pharmaceutical companies and stakeholders over the years [78]. In addition to fully academic trials, EORTC collaborates with partners within partially industry-supported trials and fully industry-supported trials, where EORTC maintains a set of “principles of independence” to safeguard scientific integrity (https://www.eortc.org/strategic-overview/). EORTC suggests an additional approach for the future of clinical research within the academic setting, where nontraditional partnerships could be developed (e.g., with regulatory, patient organizations, health technology assessment bodies) to conduct studies with a patient-centered approach (and not investigational product-centered).
A recent initiative such as “bigpicture”, which aims to set up a central repository of digital pathology images in order to boost the development of AI and subsequently allow for an exchange of (in silico) data between different research facilities and funding partners is another possibility to create new research projects (https://bigpicture.eu). Bigpicture is a public–private partnership funded by the EU Innovative Medicines Initiative and collaborates with a variety of partners ranging from academic institutions to pharmaceutical companies.
Another approach is conducting academic research within the framework of EU consortia: Another recent example is the DEFINITVE project (diagnostic HER2DX®-guided treatment for patients with early-stageHER2-positive breast cancer) under the sponsorship of the Hospital Clínic de Barcelona (FRCB-HCB) that will be funded by the European Commission, and constitutes a collaborative effort of many hospitals, research groups, universities, and cancer care organizations in several European countries (https://www.clinicbarcelona.org/en/news/a-european-project-led-by-idibaps-will-test-a-diagnostic-genomic-test-for-breast-cancer-to-improve-patients-quality-of-life).
At ABCSG, we have additionally been working on the development of a strategic compromise in the field of tension between academic and commercial control of clinical research: the “transparency model” is aiming to combine the best knowledge in trial conduct from academic and industry partners ruled by a governance process at eye level. All central study plans, guidelines, and processes are developed together and database access is regulated strictly on an expertise role-based model instead of a company model irrespective of who technically holds the database. Independent academic validation of data cleaning procedures as well as statistical analyses results, together with a clear commitment to “delivery of results in time”, form the central cornerstone of this new collaboration model. Robust, controlled procedures, blinded or masked where applicable, and technical restrictions outrule any operational or statistical bias throughout the trial conduct. Together with the delegation of major operational tasks to collaborative academic networks, this model could serve as role model for future industry–academia collaboration. We still need to prove the success—but we truly believe that such operational and governance middle ground can combine the best of all worlds—and eventually serve patients’ interests best.
Take-home message
Clinical trials remain the mainstay of knowledge generation in medicine. Despite challenges such as increasing (regulatory) requirements in clinical trials for all involved stakeholders, we examine how new technologies, integrative approaches as well as forward-thinking trial designs can positively contribute to the evolution of clinical research for the benefit of patients.
References
Li BT, Daly B, Gospodarowicz M, et al. Reimagining patient-centric cancer clinical trials: a multi-stakeholder international coalition. Nat Med. 2022;28(4):620–6. https://doi.org/10.1038/s41591-022-01775-6.
Ford I, Norrie J. Pragmatic Trials. N Engl J Med. 2016;375(5):454–63. https://doi.org/10.1056/NEJMra1510059.
Patsopoulos NA. A pragmatic view on pragmatic trials. Dialogues Clin Neurosci. 2011;13(2):217–24. https://doi.org/10.31887/DCNS.2011.13.2/npatsopoulos.
Harmon DM, Noseworthy PA, Yao X. The digitization and decentralization of clinical trials. Mayo Clin Proc. 2023;98(10):1568–78. https://doi.org/10.1016/j.mayocp.2022.10.001.
da Fonseca MH, Kovaleski F, Picinin CT, Pedroso B, Rubbo P. E‑health practices and technologies: a systematic review from 2014 to 2019. Healthcare. 2021; https://doi.org/10.3390/healthcare9091192.
Steger GG, Galid A, Gnant M, et al. Pathologic complete response with six compared with three cycles of neoadjuvant epirubicin plus docetaxel and granulocyte colony-stimulating factor in operable breast cancer: results of ABCSG-14. J Clin Oncol. 2007;25(15):2012–8. https://doi.org/10.1200/jco.2006.09.1777.
Dubsky PC, Jakesz R, Mlineritsch B, et al. Tamoxifen and anastrozole as a sequencing strategy: a randomized controlled trial in postmenopausal patients with endocrine-responsive early breast cancer from the Austrian Breast and Colorectal Cancer Study Group. J Clin Oncol. 2012;30(7):722–8. https://doi.org/10.1200/jco.2011.36.8993.
Jakesz R, Greil R, Gnant M, et al. Extended adjuvant therapy with anastrozole among postmenopausal breast cancer patients: results from the randomized Austrian Breast and Colorectal Cancer Study Group Trial 6a. J Natl Cancer Inst. 2007;99(24):1845–53. https://doi.org/10.1093/jnci/djm246.
Gnant M, Pfeiler G, Steger GG, et al. Adjuvant denosumab in postmenopausal patients with hormone receptor-positive breast cancer (ABCSG-18): disease-free survival results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019;20(3):339–51. https://doi.org/10.1016/s1470-2045(18)30862-3.
Gnant M, Pfeiler G, Dubsky PC, et al. Adjuvant denosumab in breast cancer (ABCSG-18): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. 2015;386(9992):433–43. https://doi.org/10.1016/s0140-6736(15)60995-3.
Bartsch R, Singer CF, Pfeiler G, et al. Conventional versus reverse sequence of neoadjuvant epirubicin/cyclophosphamide and docetaxel: sequencing results from ABCSG-34. Br J Cancer. 2021;124(11):1795–802. https://doi.org/10.1038/s41416-021-01284-2.
Singer CF, Pfeiler G, Hubalek M, et al. Efficacy and safety of the therapeutic cancer vaccine tecemotide (L-BLP25) in early breast cancer: Results from a prospective, randomised, neoadjuvant phase II study (ABCSG 34). Eur J Cancer. 2020;132:43–52. https://doi.org/10.1016/j.ejca.2020.03.018.
Gnant M, Mlineritsch B, Schippinger W, et al. Endocrine therapy plus zoledronic acid in premenopausal breast cancer. N Engl J Med. 2009;360(7):679–91. https://doi.org/10.1056/NEJMoa0806285.
Pötter R, Gnant M, Kwasny W, et al. Lumpectomy plus tamoxifen or anastrozole with or without whole breast irradiation in women with favorable early breast cancer. Int J Radiat Oncol Biol Phys. 2007;68(2):334–40. https://doi.org/10.1016/j.ijrobp.2006.12.045.
Geyer CE Jr., Garber JE, Gelber RD, et al. Overall survival in the OlympiA phase III trial of adjuvant olaparib in patients with germline pathogenic variants in BRCA 1/2 and high-risk, early breast cancer. Ann Oncol. 2022;33(12):1250–68. https://doi.org/10.1016/j.annonc.2022.09.159.
Schmid M, Jakesz R, Samonigg H, et al. Randomized trial of tamoxifen versus tamoxifen plus aminoglutethimide as adjuvant treatment in postmenopausal breast cancer patients with hormone receptor-positive disease: Austrian breast and colorectal cancer study group trial 6. J Clin Oncol. 2003;21(6):984–90. https://doi.org/10.1200/jco.2003.01.138.
Jakesz R, Hausmaninger H, Kubista E, et al. Randomized adjuvant trial of tamoxifen and goserelin versus cyclophosphamide, methotrexate, and fluorouracil: evidence for the superiority of treatment with endocrine blockade in premenopausal patients with hormone-responsive breast cancer—Austrian Breast and Colorectal Cancer Study Group Trial 5. J Clin Oncol. 2002;20(24):4621–7. https://doi.org/10.1200/jco.2002.09.112.
Jakesz R, Samonigg H, Gnant M, et al. Very low-dose adjuvant chemotherapy in steroid receptor negative stage I breast cancer patients. Austrian Breast Cancer Study Group. Eur J Cancer. 1998;34(1):66–70. https://doi.org/10.1016/s0959-8049(97)10010-7.
Jakesz R, Hausmaninger H, Haider K, et al. Randomized trial of low-dose chemotherapy added to tamoxifen in patients with receptor-positive and lymph node-positive breast cancer. J Clin Oncol. 1999;17(6):1701–9. https://doi.org/10.1200/jco.1999.17.6.1701.
Jakesz R, Jonat W, Gnant M, et al. Switching of postmenopausal women with endocrine-responsive early breast cancer to anastrozole after 2 years’ adjuvant tamoxifen: combined results of ABCSG trial 8 and ARNO 95 trial. Lancet. 2005;366(9484):455–62. https://doi.org/10.1016/s0140-6736(05)67059-6.
Jakesz R, Samonigg H, Gnant M, et al. Significant increase in breast conservation in 16 years of trials conducted by the Austrian Breast & Colorectal Cancer Study Group. Ann Surg. 2003;237(4):556–64. https://doi.org/10.1097/01.Sla.0000059990.43981.4e.
Gnant MF, Blijham G, Reiner A, et al. DNA ploidy and other results of DNA flow cytometry as prognostic factors in operable breast cancer: 10 year results of a randomised study. Eur J Cancer. 1992;28(2–3):711–6. https://doi.org/10.1016/s0959-8049(05)80132-7.
Gomez LE, Bernet P. Diversity improves performance and outcomes. J Natl Med Assoc. 2019;111(4):383–92. https://doi.org/10.1016/j.jnma.2019.01.006.
Clark LT, Watkins L, Pina IL, et al. Increasing diversity in clinical trials: overcoming critical barriers. Curr Probl Cardiol. 2019;44(5):148–72. https://doi.org/10.1016/j.cpcardiol.2018.11.002.
Bottern J, Stage TB, Dunvald AD. Sex, racial, and ethnic diversity in clinical trials. Clin Transl Sci. 2023;16(6):937–45. https://doi.org/10.1111/cts.13513.
Corneli A, Hanlen-Rosado E, McKenna K, et al. Enhancing diversity and inclusion in clinical trials. Clin Pharmacol Ther. 2023;113(3):489–99. https://doi.org/10.1002/cpt.2819.
McGregor B, Belton A, Henry TL, Wrenn G, Holden KB. Improving behavioral health equity through cultural competence training of health care providers. Ethn Dis. 2019;29(Suppl 2):359–64. https://doi.org/10.18865/ed.29.S2.359.
Richards R, Kinnersley P, Brain K, McCutchan G, Staffurth J, Wood F. Use of mobile devices to help cancer patients meet their information needs in non-inpatient settings: systematic review. JMIR Mhealth Uhealth. 2018;6(12):e10026. https://doi.org/10.2196/10026.
Majumder A, Sen D. Artificial intelligence in cancer diagnostics and therapy: current perspectives. Indian J Cancer. 2021;58(4):481–92. https://doi.org/10.4103/ijc.IJC_399_20.
Rivera CS, Liu X, Chan AW, et al. Guidelines for clinical trial protocols for interventions involving artificial intelligence: the SPIRIT-AI extension. Nat Med. 2020;26(9):1351–63. https://doi.org/10.1038/s41591-020-1037-7.
Filipits M, Rudas M, Singer CF, et al. ESR1, PGR, ERBB2, and MKi67 mRNA expression in postmenopausal women with hormone receptor-positive early breast cancer: results from ABCSG Trial 6. Esmo Open. 2021;6(4):100228. https://doi.org/10.1016/j.esmoop.2021.100228.
Filipits M, Rudas M, Kainz V, et al. The OncomasTR test predicts distant recurrence in estrogen receptor-positive, HER2-negative early-stage breast cancer: a validation study in ABCSG trial 8. Clin Cancer Res. 2021;27(21):5931–8. https://doi.org/10.1158/1078-0432.Ccr-21-1023.
Filipits M, Dubsky P, Rudas M, et al. Prediction of distant recurrence using endopredict among women with ER(+), HER2(−) node-positive and node-negative breast cancer treated with endocrine therapy only. Clin Cancer Res. 2019;25(13):3865–72. https://doi.org/10.1158/1078-0432.Ccr-19-0376.
Filipits M, Dafni U, Gnant M, et al. Association of p27 and Cyclin D1 expression and benefit from adjuvant trastuzumab treatment in HER2-positive early breast cancer: a TransHERA study. Clin Cancer Res. 2018;24(13):3079–86. https://doi.org/10.1158/1078-0432.Ccr-17-3473.
Bago-Horvath Z, Rudas M, Singer CF, et al. Predictive value of molecular subtypes in premenopausal women with hormone receptor-positive early breast cancer: results from the ABCSG trial 5. Clin Cancer Res. 2020;26(21):5682–8. https://doi.org/10.1158/1078-0432.Ccr-20-0673.
Rudas M, Lehnert M, Huynh A, et al. Cyclin D1 expression in breast cancer patients receiving adjuvant tamoxifen-based therapy. Clin Cancer Res. 2008;14(6):1767–74. https://doi.org/10.1158/1078-0432.Ccr-07-4122.
Gnant M, Sestak I, Filipits M, et al. Identifying clinically relevant prognostic subgroups of postmenopausal women with node-positive hormone receptor-positive early-stage breast cancer treated with endocrine therapy: a combined analysis of ABCSG‑8 and ATAC using the PAM50 risk of recurrence score and intrinsic subtype. Ann Oncol. 2015;26(8):1685–91. https://doi.org/10.1093/annonc/mdv215.
Tendl-Schulz KA, Rössler F, Wimmer P, et al. Factors influencing agreement of breast cancer luminal molecular subtype by Ki67 labeling index between core needle biopsy and surgical resection specimens. Virchows Arch. 2020;477(4):545–55. https://doi.org/10.1007/s00428-020-02818-4.
Filipits M, Nielsen TO, Rudas M, et al. The PAM50 risk-of-recurrence score predicts risk for late distant recurrence after endocrine therapy in postmenopausal women with endocrine-responsive early breast cancer. Clin Cancer Res. 2014;20(5):1298–305. https://doi.org/10.1158/1078-0432.Ccr-13-1845.
Gnant M, Filipits M, Greil R, et al. Predicting distant recurrence in receptor-positive breast cancer patients with limited clinicopathological risk: using the PAM50 Risk of Recurrence score in 1478 postmenopausal patients of the ABCSG-8 trial treated with adjuvant endocrine therapy alone. Ann Oncol. 2014;25(2):339–45. https://doi.org/10.1093/annonc/mdt494.
Filipits M, Rudas M, Jakesz R, et al. A new molecular predictor of distant recurrence in ER-positive, HER2-negative breast cancer adds independent information to conventional clinical risk factors. Clin Cancer Res. 2011;17(18):6012–20. https://doi.org/10.1158/1078-0432.Ccr-11-0926.
Filipits M, Rudas M, Heinzl H, et al. Low p27 expression predicts early relapse and death in postmenopausal hormone receptor-positive breast cancer patients receiving adjuvant tamoxifen therapy. Clin Cancer Res. 2009;15(18):5888–94. https://doi.org/10.1158/1078-0432.Ccr-09-0728.
Vargas G, Bouchet M, Bouazza L, et al. ERRα promotes breast cancer cell dissemination to bone by increasing RANK expression in primary breast tumors. Oncogene. 2019;38(7):950–64. https://doi.org/10.1038/s41388-018-0579-3.
Zhou Q, Gampenrieder SP, Frantal S, et al. Persistence of ctDNA in patients with breast cancer during neoadjuvant treatment is a significant predictor of poor tumor response. Clin Cancer Res. 2022;28(4):697–707. https://doi.org/10.1158/1078-0432.Ccr-21-3231.
Fitzal F, Filipits M, Fesl C, et al. PAM-50 predicts local recurrence after breast cancer surgery in postmenopausal patients with ER+/HER2− disease: results from 1204 patients in the randomized ABCSG-8 trial. Br J Surg. 2021;108(3):308–14. https://doi.org/10.1093/bjs/znaa089.
Singer CF, Holst F, Steurer S, et al. Estrogen receptor alpha gene amplification is an independent predictor of long-term outcome in postmenopausal patients with endocrine-responsive early breast cancer. Clin Cancer Res. 2022;28(18):4112–20. https://doi.org/10.1158/1078-0432.Ccr-21-4328.
Singer CF, Jahn SW, Rudas M, et al. Independent validation of stromal uPA in ABCSG-08: Level 1b evidence for the prognostic value of uPA immunohistochemistry. Breast. 2022;64:127–33. https://doi.org/10.1016/j.breast.2022.05.003.
ICH. Harmonised Guideline. Addendum on estimands and sensitivity analysis in clinical trials to the guideline on statistical principles for clinical trials E9(R1). International Council for Harmonisation of Technical Requirements for Pharmaceuticals For Human Use. (https://database.ich.org/sites/default/files/E9-R1_Step4_Guideline_2019_1203.pdf ).
Casey M, Degtyarev E, Lechuga MJ, et al. Estimand framework: Are we asking the right questions? A case study in the solid tumor setting. Pharm Stat. 2021;20(2):324–34. https://doi.org/10.1002/pst.2079.
Rufibach K. Treatment effect quantification for time-to-event endpoints-Estimands, analysis strategies, and beyond. Pharm Stat. 2019;18(2):145–65. https://doi.org/10.1002/pst.1917.
Young JG, Stensrud MJ, Tchetgen Tchetgen EJ, Hernan MA. A causal framework for classical statistical estimands in failure-time settings with competing events. Stat Med. 2020;39(8):1199–236. https://doi.org/10.1002/sim.8471.
Darken P, Nyberg J, Ballal S, Wright D. The attributable estimand: A new approach to account for intercurrent events. Pharm Stat. 2020;19(5):626–35. https://doi.org/10.1002/pst.2019.
Manitz J, Kan-Dobrosky N, Buchner H, et al. Estimands for overall survival in clinical trials with treatment switching in oncology. Pharm Stat. 2022;21(1):150–62. https://doi.org/10.1002/pst.2158.
Buyse M. Generalized pairwise comparisons of prioritized outcomes in the two-sample problem. Stat Med. 2010;29(30):3245–57. https://doi.org/10.1002/sim.3923.
Cantagallo E, De Backer M, Kicinski M, et al. A new measure of treatment effect in clinical trials involving competing risks based on generalized pairwise comparisons. Biom J. 2021;63(2):272–88. https://doi.org/10.1002/bimj.201900354.
Mao L. On restricted mean time in favor of treatment. Biometrics. 2023;79(1):61–72. https://doi.org/10.1111/biom.13570.
Peron J, Buyse M, Ozenne B, Roche L, Roy P. An extension of generalized pairwise comparisons for prioritized outcomes in the presence of censoring. Stat Methods Med Res. 2018;27(4):1230–9. https://doi.org/10.1177/0962280216658320.
Péron J, Roy P, Ozenne B, Roche L, Buyse M. The net chance of a longer survival as a patient-oriented measure of treatment benefit in randomized clinical trials. JAMA Oncol. 2016;2(7):901–5. https://doi.org/10.1001/jamaoncol.2015.6359.
Saad ED, Zalcberg JR, Peron J, Coart E, Burzykowski T, Buyse M. Understanding and communicating measures of treatment effect on survival: can we do better? J Natl Cancer Inst. 2018;110(3):232–40. https://doi.org/10.1093/jnci/djx179.
Buyse M, Saad ED, Burzykowski T, Péron J. Assessing treatment benefit in Immuno-oncology. Stat Biosci. 2020;12(2):83–103. https://doi.org/10.1007/s12561-020-09268-1.
Péron J, Giai J, Maucort-Boulch D, Buyse M. The benefit-risk balance of nab-Paclitaxel in metastatic pancreatic adenocarcinoma. Pancreas. 2019;48(2):275–80. https://doi.org/10.1097/mpa.0000000000001234.
Péron J, Roy P, Conroy T, et al. An assessment of the benefit-risk balance of FOLFIRINOX in metastatic pancreatic adenocarcinoma. Oncotarget. 2016;7(50):82953–60. https://doi.org/10.18632/oncotarget.12761.
Leary A, Besse B, Andre F. The need for pragmatic, affordable, and practice-changing real-life clinical trials in oncology. Lancet. 2024;403(10424):406–8. https://doi.org/10.1016/S0140-6736(23)02199-2.
Hudis CA, Barlow WE, Costantino JP, et al. Proposal for standardized definitions for efficacy end points in adjuvant breast cancer trials: the STEEP system. J Clin Oncol. 2007;25(15):2127–32. https://doi.org/10.1200/JCO.2006.10.3523.
Tolaney SM, Garrett-Mayer E, White J, et al. Updated standardized definitions for efficacy end points (STEEP) in Adjuvant breast cancer clinical trials: STEEP version 2.0. J Clin Oncol. 2021;39(24):2720–31. https://doi.org/10.1200/JCO.20.03613.
Deltuvaite-Thomas V, Verbeeck J, Burzykowski T, et al. Generalized pairwise comparisons for censored data: An overview. Biom J. 2023;65(2):e2100354. https://doi.org/10.1002/bimj.202100354.
Roustit M, Demarcq O, Laporte S, et al. Platform trials. Therapie. 2023;78(1):29–38. https://doi.org/10.1016/j.therap.2022.12.003.
Parmar MK, Barthel FM, Sydes M, et al. Speeding up the evaluation of new agents in cancer. J Natl Cancer Inst. 2008;100(17):1204–14. https://doi.org/10.1093/jnci/djn267.
Spreafico A, Hansen AR, Razak AAR, Bedard PL, Siu LL. The future of clinical trial design in oncology. Cancer Discov. 2021;11(4):822–37. https://doi.org/10.1158/2159-8290.CD-20-1301.
Kelley WN, Randolph MA. From the institute of medicine. JAMA. 1995;273(1):12. https://doi.org/10.1001/jama.273.1.12.
Nghiem VT, Vaidya R, Lyman GH, Hershman DL, Ramsey SD, Unger JM. Economic evaluations in national cancer institute-sponsored network cancer clinical trials. Value Health. 2020;23(12):1653–61. https://doi.org/10.1016/j.jval.2020.08.2095.
Nghiem VT, Vaidya R, Unger JM. Patterns of scientific and clinical impact in cancer randomized clinical trials. JAMA Netw Open. 2022;5(6):e2219657. https://doi.org/10.1001/jamanetworkopen.2022.19657.
Ralaidovy AH, Adam T, Boucher P. Resource allocation for biomedical research: analysis of investments by major funders. Health Res Policy Syst. 2020;18(1):20. https://doi.org/10.1186/s12961-020-0532-0.
Gnant M, Dueck AC, Frantal S, et al. Adjuvant palbociclib for early breast cancer: the PALLAS trial results (ABCSG-42/AFT-05/BIG-14-03). J Clin Oncol. 2022;40(3):282–93. https://doi.org/10.1200/jco.21.02554.
Piccart M, Goldhirsch A, Straehle C. The Breast International Group. a new spirit of collaboration in breast cancer research for the new millennium. Eur J Cancer. 2000;36(14):1733–6. https://doi.org/10.1016/s0959-8049(00)00164-7.
Piccart MJ, Gingras I. Breast cancer in 2015: Academic research sheds light on issues that matter to patients. Nat Rev Clin Oncol. 2016;13(2):67–8. https://doi.org/10.1038/nrclinonc.2015.236.
Riaz H, Raza S, Khan MS, Riaz IB, Krasuski RA. Impact of funding source on clinical trial results including cardiovascular outcome trials. Am J Cardiol. 2015;116(12):1944–7. https://doi.org/10.1016/j.amjcard.2015.09.034.
Broes S, Saesen R, Lacombe D, Huys I. Past, Current, and Future Cancer Clinical Research Collaborations: The Case of the European Organisation for Research and Treatment of Cancer. Clin Transl Sci. 2021;14(1):47–53. https://doi.org/10.1111/cts.12863.
Acknowledgements
On behalf of the Austrian Breast and Colorectal Cancer Study Group, the authors would like to express their gratitude first and foremost to all patients and study participants as well as their families who enable and support clinical research, and thereby build the basis for all scientific endeavors in the field of clinical oncology and translational research. We appreciate the decade-long enthusiasm, dedication, and cooperation of caregivers and their institutions in Austria and elsewhere in the world. Furthermore, we thank all cooperative study groups and collaboration partners, including the pharmaceutical and biotech industries, without whom successful collaboration and funding of clinical trials would not be feasible. Also, we are grateful to a wonderful team of ABCSG that has helped to make this a role-model enterprise for our country.
Funding
Open access funding provided by Medical University of Vienna.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
M. Gnant, M. Gili, M. Schwarz, C. Fesl, D. Hlauschek, A. Jallitsch-Halper and H. Fohler declare that there is no potential conflict of interest with respect to the submitted manuscript, except that they are employees of ABCSG and ABCSG GmbH, respectively. Outside the submitted work, M. Gnant reports personal fees/travel support from Amgen, AstraZeneca, DaiichiSankyo, EliLilly, EPG Health (IQVIA), Menarini-Stemline, MSD, Novartis, PierreFabre, Veracyte; an immediate family member is employed by Sandoz.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gnant, M., Gili, M., Schwarz, M. et al. The future of clinical trials—goals, ideas, and discussion. memo (2024). https://doi.org/10.1007/s12254-024-00969-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12254-024-00969-7