
Abstract
Transcriptomic changes in plants during drought stress give insights into the mechanisms with which plants stabilise their metabolic processes in order to cope with the drought condition. In potato, understanding such drought-induced transcriptomic changes is critical because prolonged field drought interferes with tuber formation and bulking period of potato development, which eventually affects yield. We hypothesised that phenotypic drought responses of potato genotypes may be linked to differences in transcriptomic changes. Using an RNA sequencing approach, we investigated such transcriptomic changes in leaves of three cultivars (Biogold, Hansa and Lady Rosetta) under drought. We found more differentially expressed genes (DEGs) in the tolerant cultivars, Lady Rosetta and Biogold, than in the sensitive cultivar (Hansa). The differential gene expression trend reflected the phenotypic drought responses of the cultivars. For instance, we found in both Biogold and Lady Rosetta but not in Hansa, an upregulation of genes involved in carbohydrate metabolism (e.g., Alpha-glucosidase), flavonoid biosynthesis (e.g., Flavanone 3 beta-hydroxylase), lipid biosynthesis/transfer (e.g., nonspecific Lipid Transfer Proteins), heat shock proteins and secondary metabolites like phenolics and lignins. Furthermore, a prolonged drought stress resulted in reduced DEGs in Biogold and Hansa, but not in Lady Rosetta that also maintained its tuber yield under such prolonged stress suggesting a more robust drought tolerance. Our findings suggest that a synergistic expression of genes involved in several different aspects of drought response is required in order to obtain a robust tolerance.
Similar content being viewed by others

Introduction
Drought stress is a major problem that reduces agricultural productivity. Its effects on plants primarily begin with changes in the expression of genes involved in survival or adaptation to the stress. Drought is not a single or simple stress, but a complex stress that often predisposes the plant to other stresses (Whitmore and Whalley 2009). The uncertainties of drought timing, duration and severity, which are heightened by climate change, are part of the complexity in understanding drought stress (Hosseinizadeh et al. 2015; Jenkins and Warren 2015). However, the major complexity of drought stress has been associated with the genomic composition of the plant (Blum 2011). In cultivated potato, this includes the tetraploid genetics that makes the breeding for drought tolerance difficult due to the complex inheritance of traits after genetic crosses. Also, Anithakumari et al. (2011) demonstrated in diploid potato population that drought is controlled by many loci on the genome that individually make small contributions to drought responses. Therefore, mapping drought tolerance QTLs in the genome of cultivated potato may be quite complex and hardly exhaustive. In order to understand the basis of the drought response mechanisms in potato as well as in other crops, molecular research approaches have been adopted. These have resulted in the identification of genes that regulate various aspects of drought response (Vasquez-Robinet et al. 2008; Dongjin et al. 2011; Xu et al. 2014; Obidiegwu et al. 2015; Szalonek et al. 2015; Gazendam et al. 2016). However, the regulation of these drought-associated genes and their interactions at the molecular level are not yet clearly understood, at least partly because these genes are part of networks and act in concert (Ambrosone et al. 2017; Pieczynski et al. 2018), and most functional annotations depend on the over-expression or knock-down of single genes. These transgenic approaches, though informative, may not enhance our knowledge of the multiple interactions that affect the functionality of the single genes. Therefore, comprehensive molecular approaches that incorporate the entire genome of the plant are being implemented in the study of molecular mechanisms. These whole-genome-based molecular investigations have been made even more feasible by advancements in sequencing technology (Heather and Chain 2016).
Through RNA sequencing, the transcripts from all active genes and their splice variants can be studied in depth (Morozova et al. 2009; Wang et al. 2009; Hoeijmakers et al. 2013; Risso et al. 2014; Li et al. 2015; Zyprych-Walczak et al. 2015; Anjum et al. 2016; Yang et al. 2016). These transcriptomic or RNA-sequencing (RNA-seq) approaches reveal Differentially Expressed Genes (DEGs) that have been previously annotated, but also facilitates the discovery of novel transcripts (Liu et al. 2015). In potato, RNA-seq has been applied in studies of the drought stress response of stolon tissues (Gong et al. 2015). The stolon tissues of a particular potato genotype (cv. Ningshu 4) in Gong et al. (2015) showed myriads of upregulated transcripts including heat shock proteins, dehydrins, aquaporin, protein phosphatases, sugar transporters and starch biosynthesis genes. In another potato transcriptomic study of drought response in a diploid mapping population using microarrays, TAS14 (an ABA-inducible dehydrin) was strongly induced by drought as a drought molecular sensor and correlated with drought recovery potential (van Muijen et al. 2016). Thus, transcriptomic analysis can be used to unveil the molecular signature involved in drought response.
In this study, we used RNA-seq analysis of mature leaf tissue to evaluate three potato cultivars with contrasting drought responses at two different time points. Our objective was to investigate the molecular basis for the contrasting drought responses we observed in the phenotypes of these cultivars. We found that drought tolerance may be linked to gene functions and the level of differential gene expression during drought.
Materials and Methods
Planting and Drought Application
Seed tubers of potato cultivars (Biogold, Hansa and Lady Rosetta) used in a previous study (Aliche et al. 2018) were obtained from the Dutch potato breeding companies, HZPC Holland BV and Meijer Potato, and pre-sprouted. Biogold and Lady Rosetta are early maturity cultivars with fairly stable yield across locations of different levels of water availability, whereas Hansa forms lots of tubers but its yield is drastically reduced under drought (Aliche et al. 2018). In other studies, Biogold and Lady Rosetta were also found to be more tolerant than Hansa (Aliche et al. 2019, 2020). In the current study, the tubers were planted in ridges under a tunnel in the field at Unifarm, Wageningen University & Research, The Netherlands. The ridges were set on clay soil and the field structure included two blocks: control (WR) and drought (DR) treatments. In each block, the cultivars were randomised as plots within the blocks. Four plants of each cultivar were planted in each plot, and two plots per block. The spacing between plants in a row was 30 cm and 75 cm between rows. Border plants were planted in between plots in each row. Flower Power sensors (ParrotR) were used to monitor environmental conditions: soil water content, temperature and light intensity. The control and drought blocks were regularly given water by sprinkler irrigation from the planting date (22 June 2016) till 2 weeks after emergence. Subsequently, irrigation was withheld from the drought block (20 days after planting till 76 days after planting). The development of drought stress was monitored using the sensors. Also, leaf tissues of the potato plants were sampled at several time-points for TAS14 mRNA expression analysis to monitor the development of drought stress in the plants (Supplementary Fig.SF1). TAS14 is known to be a good molecular indicator for drought stress from a previous study (van Muijen et al. 2016).
Data collection and Tissue Sampling
We took phenotypic measurements after 28 days of drought application (28DOD) including physiological traits (stomatal conductance and chlorophyll content) and growth/yield traits (shoot weight, tuber number and tuber weight). The physiological traits were scored on mature fully expanded leaves (5th–6th leaf from the apical meristem). At 56 days of drought application (56DOD), the plants were harvested and the fresh weights of the shoot and tuber tissues were measured. Tuber number was also counted. The shoot and tuber tissues were oven-dried at 105 °C for 72 h and the dry weights measured. At both time points, sampled leaf tissues (5th–6th leaf from the apical meristem) were immediately snap-frozen in liquid nitrogen and transferred to − 80 °C until subsequent molecular analysis. Tissues were sampled from two innermost plants per plot and pooled as a replicate.
Sample Preparation for RNA Sequencing
For RNA isolation, 50 mg of leaf tissues collected at 28DOD and 56DOD were homogenised in liquid nitrogen. RNA extraction was done by the addition of 500 µl of TRIzol for cell lysis and inhibition of RNase activity. Spin columns were used to extract the RNA as described in the Qiagen RNeasy mini kit protocol following the manufacturer’s instructions. RNA was eluted from the columns with 30 µl of RNase-free water. Qubit 2.0 Fluorometer and NanoDrop ND-1000 Spectrophotometer were used to determine the quantity and quality of total RNA. Library preparation and paired-end Illumina sequencing were outsourced to Beijing Genomics Institute (BGI), Hong Kong.
Bioinformatics and Statistical Data Analyses
Quality of the paired-end FASTQ read sequences was checked using the qualityScores function of Rsubread to determine if trimming was necessary. The buildindex function of Rsubread package was used to build genome indexes based on the potato reference genome from EnsemblPlants (SolTub_3.0) in R studio 3.6.1. The subjunc function of Rsubread package was used to align the reads to the reference genome in order to account for reads spanning exon-exon junctions. The propmapped function of Rsubread package was used to check the percentage of reads that successfully mapped to the reference genome. Using the read counts output from Rsubread, differential expression of genes was computed with the DESeq2 package in R studio 3.6.1 using a negative binomial function to model the distribution of the read counts data on the assumption of a linear mean–variance ratio. For differential expression computation, we used a design matrix that accounted for the main factors in the study (Genotype, Treatment and Time-point). We used the resulting DEGs for functional analysis in Gene Ontology and MapMan, and KEGG pathway analysis. Clustering of the samples was visualised with multi-dimensional scaling.
Results
Drought Stress Application and Assessment
Based on data from the Parrot Flower Power environmental sensors that we used to monitor soil water content percentage throughout the growth period and TAS14 molecular indicator (Supplementary Fig.SF1), the applied drought coincided in part with the tuber initiation and mostly with the tuber bulking stages of plant growth (Fig. 1). The scale of the Parrot Flower Power ranges from 8% (very dry) to 45% (saturated) soil moisture content. For our study, we defined a field drought (water limitation) at 25% moisture level. Taking into account that the Parrot sensors can only record soil moisture content till ~ 20 cm depth on furrows, drought severity perceived by roots of plants growing on ridges as a result of the receding soil moisture may have gradually progressed until the final harvest of the plants (Fig. 1). The control plants were maintained at 30% soil moisture content and above.

Soil water content percentage under control and drought treatments with an indication of stages of plant phenology (sprouting, stolon initiation, tuber formation and tuber bulking stages). Data were collected from 6 sensors (Parrot Flower Power) from which the average was calculated
Phenotypic Response to Drought
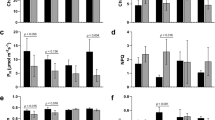
Drought stress affected plant growth with a significant shoot weight reduction in Hansa under stress (Fig. 2a) at 28DOD. Interestingly, Hansa showed a higher trend of shoot dry weight under control conditions than the other cultivars at both time points. In terms of tuber weight and tuber number, there was no drought effect at 28DOD (Fig. 2b, c), but at 56DOD, only Lady Rosetta maintained its tuber yield under stress (Fig. 2b), whereas Hansa showed a higher tuber number under control condition than the other cultivars (Fig. 2c). Stomatal conductance generally showed a reduction trend under drought, though Hansa was much more sensitive than Biogold and Lady Rosetta (Fig. 2d). Overall, Hansa was the most sensitive to drought (Fig. 2); its drought sensitivity has also been previously reported (Aliche et al. 2020). Due to the gradual drought development, plants retained their foliage throughout the experiment, although stressed plants had less canopy cover (Fig. 2e, f).

Phenotypic traits under control (WR) and stress (DR) at 28 days of drought (TDW_1, SDW_1, TBN_1 and Stomatal conductance), and 56 days of drought (TDW_2, SDW_2, TBN_2), a Tuber dry weight (TDW), b Shoot dry weight (SDW), c Tuber number (TBN), d Stomatal conductance. Error bars are standard error of the mean of four replicates. Letters represent significance of drought effects. Adjusted p values are additionally shown for stomatal conductance. e Plants growing under irrigated condition (40 days after planting), f Plants under drought stress (40 days after planting, 18 days of drought)
Overview of Sequencing Dataset
On average, 61 million paired-end reads were sequenced from each sample. Each read length was 150 bp, resulting in a mean sequencing depth of 21.8 × based on coverage computation (Lander and Waterman 1988). Quality checks of the paired-end FASTQ read sequences indicated that there was no need for trimming (data not shown). After reads alignment, 92% of the reads on average mapped to the potato reference genome (Supplementary File 1). A multidimensional scaling plot of all read counts from 42,974 genes shows the clustering of the samples according to the experimental factors, the genotypic factor having the strongest effect (Fig. 3). A phylogeny of representative gene clusters and associated treatments shows both genotypic differences and treatment effect differences within genotypes (Supplementary Fig.SF2).

a Multidimensional scaling plot of samples under control (WR) and drought (DR) conditions at 28 and 56 days of drought using read counts of 42,974 genes; b differentially upregulated genes at 28 days of drought and c differentially downregulated genes at 28 days of drought. d Differentially upregulated genes at 56 days of drought and e differentially downregulated genes at 56 days of drought
Overview of Differential Gene Expression
At the first time point, 28DOD, differential gene expression was lowest in Hansa, whereas at the second time point (56DOD), it was lowest in Biogold. There were more upregulated genes than downregulated genes at both time points in Lady Rosetta, while for Biogold, there were more downregulated genes than upregulated genes (Fig. 3). For Hansa, downregulated genes were higher than upregulated genes at 28DOD, whereas the reverse was the case at 56DOD (Fig. 3). The highest number of shared differentially expressed genes (DEGs) among the three cultivars was a downregulation of 12 genes at 28DOD (Table 1). The lists of all DEGs with their respective annotations and log2fold changes are presented in Supplementary File 2.
Gene Ontology Functional Assessment
Further, a functional assessment of the DEGs per cultivar at 28DOD using Gene Ontology (GO) enrichment at a threshold of 0.05 false discovery rate (FDR) showed that abiotic stress-related biological processes were enriched among other GO terms (Fig. 4). We investigated some of the enriched GO biological processes for which we found differences between tolerant and sensitive cultivars. The tolerant cultivars, Lady Rosetta and Biogold, showed a significant enrichment of flavonoid biosynthetic process but not the sensitive cultivar, Hansa. A follow-up RT-PCR of one of the genes contributing to this biological process, Flavanone 3 beta-hydroxylase, showed a twofold upregulation in Lady Rosetta and Biogold but not in Hansa, similar to the gene expression trend obtained from the RNAseq result (Fig. 5d). Although the GO enrichment of protein folding biological process was found in both Hansa and Lady Rosetta (Fig. 4), only in Lady Rosetta (tolerant cultivar) was an enrichment of biological processes that prevent protein impairment detected, comprising mainly DEGs that inhibit proteinase activity (data not shown). Furthermore, we found an upregulation of drought-responsive genes like aquaporin gene (water channel protein) at 28DOD, nonspecific lipid transfer protein (nsLTP) at 28DOD, ROS scavenging gene (Glutaredoxin) at 56DOD in both Biogold and Lady Rosetta but not in Hansa (Supplementary File 2). A follow-up RT-PCR experiment confirmed the RNAseq differential expression results for some of these drought-associated genes (Fig. 5e–h).

Gene Ontology (GO) biological processes significantly enriched at 28 days of drought in a Hansa, b Lady Rosetta, c Biogold, d both Lady Rosetta and Biogold. The numbers on each chart represents the number of differentially expressed genes (DEGs) associated with each GO term


Mapman display of secondary metabolites presumably produced based on upregulated DEGs at 28DOD in a Hansa, b Lady Rosetta, and c Biogold. Follow-up RT-PCR Log2fold change expression of d Flavanone-3-beta-hydroxylase at 28DOD, e Water channel protein (Aquaporin) at 28DOD, f Glutaredoxin at 56DOD, g Alpha glucosidase at 28DOD, h Protein phosphatase 2C8 at 56DOD
Pathway Analysis
We employed KEGG pathway analysis to search for pathways that were activated under stress using the DEGs. At 28DOD, biosynthesis of secondary metabolites was enriched in all three cultivars with more DEG representation found in Biogold and Lady Rosetta than the sensitive cultivar, Hansa (Table 2). Using Mapman tool, we compared the secondary metabolites that were presumably produced based on the DEGs in all three cultivars at 28DOD. The secondary metabolites supposedly produced in Hansa at 28DOD were terpenoids, whereas Biogold and Lady Rosetta may have produced terpenoids, phenylpropanoids, lignins, simple phenols like flavonoids and alkaloids (Fig. 5a–d).
Also, starch/sucrose metabolism pathways were activated in Biogold and Lady Rosetta, but not in Hansa at 28DOD (Table 2, Supplementary File 3). A follow-up RT-PCR study of Alpha glucosidase, a gene involved in starch/sucrose metabolism confirmed the RNAseq expression result for this gene (Fig. 5g).
Gene Expression Downregulation Under Drought
We investigated the GO-enriched biological processes using the downregulated DEGs at 28DOD. Whereas the tolerant cultivars, Lady Rosetta and Biogold, showed a downregulation of defence response to biotic agents, the sensitive cultivar, Hansa, showed a downregulation of processes involved in DNA-complex regulation and protein synthesis machinery (Supplementary File 4). The most downregulated DEGs at 28DOD for Biogold and Lady Rosetta were endochitinase and transcription factors, respectively, while for Hansa, a number of genes involved in cell division were among the most downregulated DEGs (Supplementary File 4).
Differential Gene Expression Overlaps Between Time Points
Using Venn diagrams, we investigated the relationships in differential gene expression between the two time points per cultivar. Unlike for Hansa and Biogold, we observed that about 50% of the upregulated DEGs at 28DOD in Lady Rosetta were also upregulated at 56DOD (Fig. 6). A further investigation of these 50% of genes (226 DEGs) that were upregulated at both times using GO analysis showed that the enriched biological processes included response to abiotic stimuli like heat, flavonoid biosynthesis, carbohydrate metabolism, cell wall modification and biogenesis (data not shown). The only gene that was upregulated at both time points in Biogold was a lipid transfer protein (PGSC0003DMG400011953) possibly involved in the deposition of wax or cutin in cell walls. Interestingly, this gene (lipid transfer protein) was also upregulated in Lady Rosetta at 28DOD but not in Hansa. Nonetheless, it was upregulated in all three cultivars at 56DOD (Supplementary File 2). On the other hand, the DEGs upregulated at both time points in Hansa included PGSC0003DMG400030008 (low temperature-induced 65 kDa protein) and PGSC0003DMG400009112 (Protein phosphatase 2C) (Supplementary File 2).

Venn diagrams showing the overlapping number of differentially expressed genes (DEGs) between two time points (28DOD and 56DOD) per cultivar a Hansa, b Lady Rosetta, c Biogold. DOD is days of drought. The signs ‘ + ’ and ‘ − ’ stand for upregulated and downregulated DEGs, respectively
Discussion
Drought stress is known to adversely affect potato growth and tuber yield (Obidiegwu et al. 2015). The observed drought response of potato is based on the molecular changes that occur after perception of drought. In this study, we have investigated drought-induced molecular changes in three genotypic backgrounds. We found a high level of variation in the number of differentially expressed genes, enriched biological processes and activated pathways among the cultivars across two time points.
A strong genotypic effect on transcriptomic changes in response to drought (Fig. 3a) suggests that different genotypes have unique ways of coping with drought. Overall, the measured phenotypic traits suggest that at 28DOD, the drought did not affect carbon partitioning to the tubers (Fig. 2b, c). However, investment in tuberisation at this time point came at the expense of shoot weight for Hansa (Fig. 2a). Aliche et al. (2020) have previously demonstrated the competition between tubers and above-ground tissues (shoot) for photosynthesised carbon, and how severe drought could arrest tuberisation thereby favouring investments in shoot. In the present study involving a milder field drought, tuberisation was not arrested and so we could observe the dynamics in carbon partitioning between tuber and shoot, in which case the most sensitive cultivar (Hansa) was unable to balance these two opposing streams of carbon flow (Fig. 2).
At the transcriptomic level, interestingly, our findings suggest that drought tolerance requires the initiation of several protective but also resilience and survival molecular mechanisms in addition to the perception of the drought. The several folds upregulation of dehydrin (environmental stress-inducible gene TAS14—Supplementary Fig.SF1), low temperature-induced 65 kDa protein and heat shock proteins in Hansa (Supplementary File 2) were not sufficient to attain drought tolerance in this cultivar. In the tolerant cultivars, on the other hand, the upregulation of genes involved in carbon metabolism, secondary metabolites, detoxification and lipid transfer proteins, in addition to the drought perception genes (TAS14) and heat shock proteins (Supplementary File 2, Supplementary Fig.SF1), most likely have contributed to the drought tolerance of these two cultivars. In a previous study, genome-wide comparative RNAseq analyses were used to identify potato genes and their homologues in Arabidopsis that are evolutionarily conserved in drought response (Pieczynski et al. 2018). The authors found seven genes involved in different aspects of plant molecular mechanisms including carbohydrate transport, MAPKKK and nonspecific lipid transfer protein, suggesting that the evolutionary interplay between potato and drought stress requires a synergy of several molecular pathways. In our study, we observed such synergistic mechanisms of different pathways in the tolerant cultivars as shown in the expression of genes involved in carbohydrate metabolism and nonspecific lipid transfer proteins, which were not upregulated in the drought-sensitive cultivar (Supplementary File 2). The non-induction of these essential genes in the sensitive cultivar must have resulted in non-maintenance of turgor, absence of waterproof lipid barriers that minimise water loss and flavonoids, and limited carbohydrates metabolism leading to the observed poor biomass partitioning. Regarding carbon partitioning, a previous study has shown that the downregulation of genes involved in sucrose metabolism under drought could contribute to drought sensitivity (Evers et al. 2010). In our study, therefore, we propose that the enrichment of sucrose metabolism in Lady Rosetta and Biogold must have significantly aided the maintenance of biomass under stress in contrast to Hansa (Fig. 2a).
In addition to the importance of primary metabolites like sugars and lipids under drought conditions, secondary metabolites may be equally important, though understudied. According to Niinemets (2016), secondary metabolites may be critical for an efficient recovery from drought stress or plant’s resilience to subsequent occurrence of other stresses. The interaction between drought stress and the production of secondary metabolites has been elaborated in another study (Selmar and Kleinwächter 2013), suggesting that drought-induced carbon-uptake shortage which results in superabundance of reduction equivalents (NADPH + H+) tends to favour metabolic processes that use up such reduction equivalents so as to inhibit oxidative stress. This in turn induces the production of reduced compounds including secondary metabolites. In our study, drought tolerance tends to be associated with upregulation of genes involved in the synthesis of secondary metabolites like lignin production and phenolics. There is sufficient evidence supporting the protective role of lignin under drought stress as it has been shown in rice with the respective overexpression of OsCCR10 and SiMYB56 in different studies, that drought-induced lignin production resulted in drought tolerance (Choi 2019; Xu et al. 2020). Lignin is essential in maintaining the integrity of plant vascular tissues to ensure efficient water transport to tissues that need it most during stress (Malavasi et al. 2016). Likewise, the essential roles of plant phenolics (e.g., phenylpropanoids, flavonoids and phenols) in drought tolerance, like scavenging reactive oxygen species among other functions, have been reported in several studies (Sharma et al. 2019;Kumar et al. 2020; Li et al. 2020). Taken together, our results show that the supposed higher production of secondary metabolites in Lady Rosetta and Biogold than Hansa (Fig. 5) may have contributed to the differences in tolerance that we observed between this set of cultivars (Fig. 2).
The robustness of drought tolerance may be negatively impacted by a long duration of drought stress that also often heightens drought severity. In our study, robustness of drought tolerance was tested at 56DOD and tuber yield significantly distinguished Lady Rosetta as robustly tolerant when compared to Biogold and Hansa (Fig. 2b). Also, the higher number of DEGs at 56DOD than 28DOD observed in Lady Rosetta suggests a relatively more robust molecular resilience for likely increase in drought severity than the other cultivars. It has recently been shown in another transcriptomic study using a drought-tolerant potato landrace (Jancko Sisu Yari) that an increase in drought severity triggered more differential gene expression (Chen et al. 2020). This suggests in our study that the transcriptomic architecture of Lady Rosetta may have fortified it much more for drought tolerance at 56DOD than Biogold and Hansa. In fact, in addition to sustaining the upregulation of protective genes against the drought at both 28DOD and 56DOD (cell wall biogenesis, flavonoids), the genes involved in carbohydrate metabolism were upregulated at both time points in Lady Rosetta which may have contributed to its ability to keep its biomass and tuber yield at 56DOD (Fig. 2).
Conclusions
We have shown that differential gene expression under drought may contribute to phenotypic differences in drought response of potato cultivars. Drought tolerance requires the upregulation of genes involved in carbon metabolism (e.g., Alpha glucosidase), secondary metabolites (e.g., Flavanone 3 beta-hydroxylase), detoxification (e.g., Gutaredoxin) and lipid transfer proteins, in addition to drought perception and protective genes like dehydrins (TAS14) and heat shock proteins. Finally, a robust drought tolerance response would require the synergistic expression of genes involved in plant protective functions (like osmotic adjustment and cell wall maintenance) as well as carbohydrate metabolism throughout the period of drought. As a follow-up to our findings, further work will be needed to understand the extent of contribution of these various upregulated genes and pathways to drought tolerance in these genotypic backgrounds. For instance, using clones with mutations in multiple-genes (double, triple, quadruple mutants) and studying these mutation impacts on drought response.
Data Availability
RNAseq data will be archived in NCBI GenBank [Accession number: to be provided].
Code Availability
Not applicable.
References
Aliche EB, Theeuwen TPJM, Oortwijn M, Visser RGF, van der Linden CG (2020) Carbon partitioning mechanisms in potato under drought stress. Plant Physiol Biochem 146:211–219
Aliche EB, Oortwijn M, Theeuwen TPJM, Bachem CWB, van Eck HJ, Visser RGF, van der Linden CG (2019) Genetic mapping of tuber size distribution and marketable tuber yield under drought stress in potatoes. Euphytica 215:186. https://doi.org/10.1007/s10681-019-2508-0
Aliche EB, Oortwijn M, Theeuwen TPJM, Bachem CWB, Visser RGF, van der Linden CG (2018) Drought response in field grown potatoes and the interactions between canopy growth and yield. Agr Wat Mgt 206:20–30. https://doi.org/10.1016/j.agwat.2018.04.013
Ambrosone A, Batelli G, Bostan H, D’Agostino N, Chiusano ML, Perrotta G, Leone A, Grillo S, Costa A (2017) Distinct gene networks drive differential response to abrupt or gradual water deficit in potato. Gene 597:30–39. https://doi.org/10.1016/j.gene.2016.10.024
Anithakumari AM, Dolstra O, Vosman B, Visser RGF, van der Linden CG (2011) In vitro screening and QTL analysis for drought tolerance in diploid potato. Euphytica 181(3):357–369. https://doi.org/10.1007/s10681-011-0446-6
Anjum A, Jaggi S, Varghese E, Lall S, Bhowmik A, Rai A (2016) Identification of Differentially Expressed Genes in RNA-seq Data of Arabidopsis thaliana: A Compound Distribution Approach. Jour of Comp Biol 23(4):239–247. https://doi.org/10.1089/cmb.2015.0205
Blum A (2011) Drought resistance – is it really a complex trait? Fun Plant Biol 38(10):753–757. https://doi.org/10.1071/FP11101
Chen Y, Li C, Yi J, Yang Y, Lei C, Gong M (2020) Transcriptome response to drought, rehydration and re-dehydration in potato. Int J Mol Sci 21:159. https://doi.org/10.3390/ijms21010159
Choi, S (2019) Characterization of the OsCCR10, a lignin biosynthesis gene involved in rice drought tolerance mechanism (MSc. Thesis), Seoul National University, Seoul. Retrieved on 20/10/2020 from https://s-space.snu.ac.kr/bitstream/10371/161109/1/000000157214.pdf
Dongjin S, Seok-Jun M, Seyoun H, Beom-Gi K, Sang RP, Seong-Kon L, Hye-Jin Y, Hye EL, Hawk-Bin K, Dongwon B, Bu YY, Myung-Ok B (2011) Expression of StMYB1R-1, a Novel Potato Single MYB-Like Domain transcription Factor, Increases Drought Tolerance. Plant Physiol 155:421–432
Evers D, Lefèvre I, Legay S, Lamoureux D, Hausman JF, Rosales ROG, Marca LRT, Hoffmann L, Bonierbale M, Schafleitner R (2010) Identification of drought-responsive compounds in potato through a combined transcriptomic and targeted metabolite approach. J Exp Bot 61(9):2327–2343. https://doi.org/10.1093/jxb/erq060
Gazendam I, Greyling R, Laurie RN, Matsaunyane LBT, Oelofse D, Rakuambo J (2016) A transgenic approach to improve the drought tolerance of potato. Paper Presented at the Acta Horticulturae 1110:203–210. https://doi.org/10.17660/ActaHortic.2016.1110.29
Gong L, Zhang H, Gan X, Zhang L, Chen Y, Nie F, Shi L, Li M, Guo Z, Zhang G, Song Y (2015) Transcriptome Profiling of the Potato (Solanum tuberosum L.) Plant under Drought Stress and Water-Stimulus Conditions. PLoS One 10(5):e0128041. https://doi.org/10.1371/journal.pone.0128041
Harb A, Krishnan A, Ambavaram MMR, Pereira A (2010) Molecular and Physiological Analysis of Drought Stress in Arabidopsis Reveals Early Responses Leading to Acclimation in Plant Growth. Plant Physiol 154(3):1254–1271. https://doi.org/10.1104/pp.110.161752
Heather JM, Chain B (2016) The sequence of sequencers: The history of sequencing DNA. Genomics 107(1):1–8. https://doi.org/10.1016/j.ygeno.2015.11.003
Hoeijmakers WA, Bartfai R, Stunnenberg HG (2013) Transcriptome analysis using RNA-Seq. Met Mol Biol 923:221–239. https://doi.org/10.1007/978-1-62703-026-7_15
Hosseinizadeh A, SeyedKaboli H, Zareie H, Akhondali A, Farjad B (2015) Impact of climate change on the severity, duration, and frequency of drought in a semi-arid agricultural basin. Geoenv Dis 2(1):23. https://doi.org/10.1186/s40677-015-0031-8
Jenkins K, Warren R (2015) Quantifying the impact of climate change on drought regimes using the Standardised Precipitation Index. Theor and App Climatol 120(1):41–54. https://doi.org/10.1007/s00704-014-1143-x
Kumar S, Bhushan B, Wakchaure GC, Meena KK, Kumar M, Meena NL, Rane J (2020) Plant phenolics under water-deficit conditions: Biosynthesis, accumulation, and physiological roles in water stress alleviation. In: Lone R, Shuab R, Kamili A (eds) Plant Phenolics in Sustainable Agriculture. Springer, Singapore. https://doi.org/10.1007/978-981-15-4890-1_19
Lander ES, Waterman MS (1988) Genomic mapping by fingerprinting random clones: a mathematical analysis. Genomics 2(3):231–239
Li P, Piao Y, Shon HS, Ryu KH (2015) Comparing the normalization methods for the differential analysis of Illumina high-throughput RNA-Seq data. BMC Bioinf 16(1):347. https://doi.org/10.1186/s12859-015-0778-7
Li X, Zhang X, Liu G, Tang Y, Zhou C, Zhang L, Lv J (2020) The spike plays important roles in the drought tolerance as compared to the flag leaf through the phenylpropanoid pathway in wheat. Plant Physiol Biochem 152:100–111. https://doi.org/10.1016/j.plaphy.2020.05.002
Liu Y, Lin-Wang K, Deng C, Warran B, Wang L, Yu B, Yang H, Wang J, Espley RV, Zhang J, Wang D, Allan AC (2015) Comparative Transcriptome Analysis of White and Purple Potato to Identify Genes Involved in Anthocyanin Biosynthesis. PLoS ONE 10(6):e0129148
Malavasi UC, Davis AS, Malavasi MdM (2016) Lignin in woody plants under water stress: A Review. Floresta e Ambiente 23(4):589–597. https://doi.org/10.1590/2179-8087.143715
Morozova O, Hirst M, Marra MA (2009) Applications of new sequencing technologies for transcriptome analysis. Annu Rev Genomics Hum Genet 10:135–151. https://doi.org/10.1146/annurev-genom-082908-145957
Muñoz-Mayor A, Pineda B, Garcia-Abellán JO, Antón T, Garcia-Sogo B, Sanchez-Bel P, Flores FB, Atarés A, Angosto T, Pintor-Toro JA, Moreno V, Bolarin MC (2012) Overexpression of dehydrin TAS14 gene improves the osmotic stress imposed by drought and salinity in tomato. J Plant Physiol 169(5):459–468. https://doi.org/10.1016/j.jplph.2011.11.018
Niinemets Ü (2016) Uncovering the hidden facets of drought stress: secondary metabolites make the difference. Tree Physiol 36(2):129–132. https://doi.org/10.1093/treephys/tpv128
Obidiegwu JE, Bryan GJ, Jones HG, Prashar A (2015) Coping with drought: stress and adaptive responses in potato and perspectives for improvement. Front in Plant Sci 6:542. https://doi.org/10.3389/fpls.2015.00542
Pieczynski M, Wyrzykowska A, Milanowska K, Boguszewska-Mankowska D, Zagdanska B, Karlowski W, Jarmolowski A, Szweykowska-Kulinska Z (2018) Genomewide identification of genes involved in the potato response to drought indicates functional evolutionary conservation with Arabidopsis plants. Plant Biotechnol J 16(2):603–614. https://doi.org/10.1111/pbi.12800
Risso D, Ngai J, Speed TP, Dudoit S (2014) Normalization of RNA-seq data using factor analysis of control genes or samples. Nat Biotech 32(9):896–902
Selmar D, Kleinwächter M (2013) Stress Enhances the Synthesis of Secondary Plant Products: The Impact of Stress-Related Over-Reduction on the Accumulation of Natural Products. Plant and Cell Physiol 54(6):817–826. https://doi.org/10.1093/pcp/pct054
Sharma A, Shahzad B, Rehman A, Bhardwaj R, Landi M, Zheng B (2019) Response of phenylpropanoid pathway and the role of polyphenols in plants under abiotic stress. Molecules 24(13):2452. https://doi.org/10.3390/molecules24132452
Szalonek M, Sierpien B, Rymaszewski W, Gieczewska K, Garstka M, Lichocka M, Sass L, Paul K, Vass I, Vankova R, Dobrev P, Szczesny P, Marczewski W, Krusiewicz D, Strzelczyk-Zyta D, Hennig J, Konopka-Postupolska D (2015) Potato Annexin STANN1 Promotes Drought Tolerance and Mitigates Light Stress in Transgenic Solanum tuberosum L. Plants. PLoS ONE 10(7):e0132683
Ul Haq S, Khan A, Ali M, Khattak AM, Gai WX, Zhang HX, Wei AM, Gong ZH (2019) Heat Shock Proteins: Dynamic Biomolecules to Counter Plant Biotic and Abiotic Stresses. Int J Mol Sci 20(21):5321. https://doi.org/10.3390/ijms20215321
van Muijen D, Anithakumari AM, Maliepaard C, Visser RGF, van der Linden CG (2016) Systems genetics reveals key genetic elements of drought induced gene regulation in diploid potato. Plant, Cell & Env 39(9):1895–1908. https://doi.org/10.1111/pce.12744
Vasquez-Robinet C, Mane SP, Ulanov AV, Watkinson JI, Stromberg VK, De Koeyer D, Schafleitner R, Willmot DB, Bonierbale M, Bohnert HJ, Grene R (2008) Physiological and molecular adaptations to drought in Andean potato genotypes. J Exp Bot 59(8):2109–2123. https://doi.org/10.1093/jxb/ern073
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10(1):57–63. https://doi.org/10.1038/nrg2484
Whitmore AP, Whalley WR (2009) Physical effects of soil drying on roots and crop growth. J Exp Bot 60(10):2845–2857. https://doi.org/10.1093/jxb/erp200
Xu W, Tang W, Wang C, Ge L, Sun J, Qi X, He Z, Zhou Y, Chen J, Xu Z, Ma YZ, Chen M (2020) SiMYB56 Confers Drought Stress Tolerance in Transgenic Rice by Regulating Lignin Biosynthesis and ABA Signaling Pathway. Front in Plant Sci 11:785
Xu Q, He Q, Li S, Tian Z (2014) Molecular characterization of StNAC2 in potato and its overexpression confers drought and salt tolerance. Acta Physiolog Plant 36(7):1841–1851. https://doi.org/10.1007/s11738-014-1558-0
Yang S, Mercante DE, Zhang K, Fang Z (2016) An Integrated Approach for RNA- seq Data Normalization. Cancer Info 15:129–141. https://doi.org/10.4137/cin.s39781
Zyprych-Walczak J, Szabelska A, Handschuh L, Go K, Górczak K, Klamecka K, Figlerowicz M, Siatkowski I (2015) The impact of normalization methods on RNA-seq data analysis. BioMed Res Int 2015:621690. https://doi.org/10.1155/2015/621690
Acknowledgements
We thank the following companies for their in-kind contribution and logistics support for this study: Averis Seeds, Meijer Potato, HZPC Holland B.V. and KWS POTATO
Funding
This research was funded by the TopSector (TKI Tuinbouw en Uitgangsmaterialen, Number 262), Averis Seeds, Meijer Potato, HZPC Holland B.V. and KWS POTATO.
Author information
Authors and Affiliations
Contributions
CGvdL, RGFV, CWB and EBA developed the idea for the study and wrote the manuscript; EBA, TG, IH and MO collected and processed the data; TB and EBA analysed the datasets.
Corresponding author
Ethics declarations
Ethical Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
All authors have given their consent for this publication.
Competing Interests
The authors declare that they have not conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Aliche, E.B., Gengler, T., Hoendervangers, I. et al. Transcriptomic Responses of Potato to Drought Stress. Potato Res. 65, 289–305 (2022). https://doi.org/10.1007/s11540-021-09527-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11540-021-09527-8