
Abstract
Estrogens have pleiotropic effects on many reproductive and non-reproductive tissues and organs including the mammary gland, uterus, ovaries, vagina, and endothelium. Estrogen receptor α functions as the principal mediator of estrogenic action in most of these tissues. Estetrol (E4) is a native fetal estrogen with selective tissue actions that is currently approved for use as the estrogen component in a combined oral contraceptive and is being developed as a menopause hormone therapy (MHT, also known as hormone replacement therapy). However, exogenous hormonal treatments, in particular MHTs, have been shown to promote the growth of preexisting breast cancers and are associated with a variable risk of breast cancer depending on the treatment modality. Therefore, evaluating the safety of E4-based formulations on the breast forms a crucial part of the clinical development process. This review highlights preclinical and clinical studies that have assessed the effects of E4 and E4-progestogen combinations on the mammary gland and breast cancer, focusing in particular on the estrogenic and anti-estrogenic properties of E4. We discuss the potential advantages of E4 over current available estrogen-formulations as a contraceptive and for the treatment of symptoms due to menopause. We also consider the potential of E4 for the treatment of endocrine-resistant breast cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Estrogens are master drivers of reproductive activity in women; controlling ovulation, zygote implantation, and mammary gland development [1,2,3]. Estrogens also have pleiotropic protective effects on non-reproductive tissues and organs and help to ensure optimal health of women during their childbearing years [4,5,6,7,8,9,10]. A women’s life can be divided into five main stages based on estrogen activity: childhood, puberty, reproductive life, perimenopause and postmenopause. Estrogen levels increase during puberty until menarche, when levels stabilize. They then vary in line with the monthly menstrual cycle until late perimenopause, when estrogen levels fall and stabilize. Menopause hormone treatments (MHTs), also known as hormone replacement therapy (HRT), are the most powerful treatments available for alleviating the symptoms associated with the cessation of estrogen production by the ovaries at menopause [11, 12]. However, these estrogen-based treatments are associated with an increased risk of breast cancer and estrogens also act as growth factors in estrogen receptor-positive (ER +) breast cancers, which account for 70% of all cases [13]. Therefore, there is an unmet medical need for a new generation of MHT with an improved benefit/risk profile for breast cancer in particular.
The ideal estrogen would display the following characteristics: 1) effective on menopause symptoms (in particular hot flushes, vulvo-vaginal atrophy, and osteoporosis); 2) neutral on mammary gland and breast cancer growth; 3) neutral on endometrial hyperplasia, which increases the risk of endometrial cancer; 4) cardioprotective against atherosclerosis and thromboembolism; 5) a favorable metabolic profile.
Accumulating data support the use of estetrol (E4), a natural fetal estrogen, for such clinical applications. E4 is currently approved by the US Food and Drugs Administration and by the European Medicines Agency as the estrogen component in a combined oral contraceptive and is under development for the treatment of symptoms due to menopause. It is also being evaluated for advanced endocrine-resistant breast cancer. In this review, we aim to define the extent to which E4 fulfills ‘the ideal criteria’, focusing in particular on its effects on the mammary gland and breast cancer. We will provide a brief summary of the clinical evidence associated with breast cancer risk and the uptake of exogenous estrogen-based formulations, followed by a review of E4 biology, E4 signaling pathways, and its actions on the mammary gland and breast cancer. We will then move on to discuss the potential benefits of E4 as a contraceptive, as a treatment for symptoms due to menopause, and for the treatment of endocrine-resistant breast cancer.
Estrogen-based Formulations and Breast Cancer Risk
Estrogens are widely used as a hormonal treatment for multiple therapeutic indications, in particular for contraception and in the treatment of symptoms due to menopause. Currently, 17β-estradiol (E2) and estriol (E3) are the most widely prescribed natural estrogens in combined oral contraceptives (COCs) and in MHT [11, 12]. Synthetic estrogenic molecules such as conjugated equine estrogen, ethinyl estradiol and E2-valerate are also used [12, 14].
Over the past two decades, the safety of COCs on breast tissue has been debated in the literature and overall, the risk of COC-associated breast cancer risk appears to remain low. However, there is limited evidence available as the incidence is low, studies are difficult, and require very large cohorts of patients [15,16,17]. Interestingly, the slight increase in risk that is observed, disappears ten years or more after treatment cessation [15], highlighting that estrogens promote preexisting breast cancer cell growth rather than inducing breast carcinogenesis. Moreover, the risk of ovarian, endometrial, and colorectal cancers is reduced for women using COCs [18], and such advantages may outweigh the potential negative effect of COCs on breast cancer risk for premenopausal women.
In 2002, for the first time, the Women’s Health Initiative (WHI) study [19] reported an association between MHT use and breast cancer risk. This study had unprecedented consequences on the prescription rates of MHTs, which subsequently decreased by 30% [20]. Between 2003 and 2011, several European and American epidemiological and observational studies including, the Million Women Study, the French E3N cohort, a study of Finnish women, and the European Prospective Investigation into Cancer and Nutrition (EPIC) studies, referenced breast cancer risk as a major side effect of MHTs [19, 21,22,23,24,25]. In 2012, the Cochrane meta-analysis confirmed the pro-tumoral risk of MHT [26] and in 2019, a long-term prospective study, with a mean follow-up period of 17.6 years, showed a correlation between MHT and breast cancer-associated mortality [27]. Finally, a meta-analysis of 58 studies, including 143,887 postmenopausal women with breast cancer and 424,972 women without breast cancer, was published by the Collaborative Group on Hormonal Factors in Breast Cancer [28] and again confirmed the correlation between MHT use and breast cancer risk. However, variations in the relative risk ratio were reported depending on modality (Table 1) and all MHT formulations, with the exception of estrogenic vaginal cream, were associated with an increased risk [28]. An excess risk of breast cancer was associated with both current or recent use (1–4 years) and long-term treatment. The incidence of mammary cancers correlated with treatment duration [23, 29] and decreases progressively after treatment cessation [21, 29]. The association between breast cancer and MHT use was higher for estrogen-progestogen combinations compared to estrogen-only formulations or placebo [12, 21, 24,25,26, 28]. Combinations with natural progesterone and dydrogesterone appeared to be safer [22] than those combined with synthetic progestins such as norethisterone acetate and medroxy-progesterone acetate (MPA), which potentiated the breast cancer risk [23, 25]. Both estrogenic and combined MHTs preferentially induced ER + breast cancer [28]. These observations support a role for MHTs in the potentiation of preexisting breast cancer cells rather than in the induction of carcinogenesis. In summary, the risk of breast cancer in MHT users was highest for oral estrogen-progestogen formulations used for more than 5 years.
Together these data suggest that, if estrogens promote ER + breast cancer growth, exogenous administration of estrogen combined with a progestogen as a MHT in postmenopausal women could increase the risk of breast cancer. In western countries, MHTs are prescribed to almost 12 million women, highlighting the clinical need [28]. The following section aims to provide a critical overview of E4 and to define the potential advantages of E4 over conventional estrogens in the development of a new generation of MHT.
Estetrol Biology
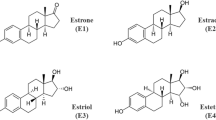
There are four natural estrogens synthetized in humans, estrone (E1), E2, E3 and E4 (Fig. 1). E4 was discovered in 1965 by the team of Diczfalusy [30]. It is produced exclusively during pregnancy by the liver in both male and female fetuses [30, 31], through 15α- and/or 16α-hydroxylation of E2 or E3 [31]. E4 is detected from the 9th week of gestation in maternal urine and from the 20th week in the maternal plasma. Maternal plasma rates increase during pregnancy and reach 1 ng/ml (3 nM) by the second trimester. E4 fetal plasma levels at term are 12 times higher than those of the mother [32, 33]. Despite work in the 80 s that evaluated maternal E4 level as an index of pregnancy complications and fetus well-being [32, 34, 35], the physiological role of E4 during pregnancy still remains undefined.

Biochemical structures of natural estrogens. Schematic representation of endogenous estrogens, estrone (E1), 17β-estradiol (E2), estriol (E3) and estetrol (E4). Structural images were uploaded from ChemSpider (www.chemspider.com) with permission
In humans, E4 is characterized by a high oral bioavailability of 90% [36], compared to 10% for E2. The human half-life of E4 is 28–32 h [36], compared to 90 min for E2 [14]. In rodents, the bioavailability of E4 is approximately 70% and its half-life is between 2 and 3 h [37]. Both absorption and oral bioavailability are dose-dependent and interindividual plasma variations after oral administration are low [36]. Taken together, the pharmacokinetic parameters of E4 make it suitable for oral use. The hepatic metabolism of E4 is slow in both humans and rodents and is similar across the two species. E4 metabolites are produced through conjugation, mainly methylation, (de)hydroxylation, glucuronidation and sulfation, they are inactive [32, 38] and are rapidly excreted in urine [32, 38,39,40]. The principal urine metabolite is the Ring D-monoglucuronide, but E4 can also be excreted in an unconjugated form that cannot be reciprocally converted back into E2 or E3 [35, 36].
E4 is selective for the estrogen receptors (estrogen receptor alpha [ERα] and estrogen receptor beta [ERβ]) and binds poorly to other nuclear receptors even at very high concentrations [38]. Although the binding affinity of E4 for both ERα and ERβ is moderate in comparison to E2 [38, 41] (Table 2), its binding affinity for ERα is almost 5 times higher than that of ERβ (Table 2). The interaction of E4 with ERα, at the ligand binding domain is similar to E2 [41]. There is currently no data available on potential binding of E4 to G-coupled protein estrogen receptor (GPER).
Estetrol Signaling Pathways
In transgenic mouse studies, ERα has been shown to mediate most of the estrogenic actions in organs including the brain, endothelium, mammary gland, vagina, and uterus. ERα has also been shown to control processes such as atheroprotection, vasodilatation, nitric oxide synthesis, endothelial healing, and bone demineralization, and is also involved in the prevention of type-2 diabetes [7, 42,43,44,45,46,47,48,49]. Two distinct signaling pathways are associated with ERα activation (reviewed in [50]): the genomic/nuclear pathway [51] and the non-genomic/extra-nuclear/membrane-initiated steroid signaling (MISS) pathway [50, 52]. The genomic pathway is associated with the transcriptional activity of the nuclear form of the receptor, while the MISS pathway is induced by the membrane-anchored form of ERα or GPER and leads to rapid activation of intracellular signaling cascades (Fig. 2).

ERα signaling pathways. Schematic representation of the genomic pathway and the MISS pathway associated with E2-induced ERα signaling. Abbreviations: S118p, Serine 118 phosphorylated ERα; Co-Reg, co-regulators; TF, transcription factors
The signaling pathways associated with E4 have been extensively studied in the endothelium in vivo and have been compared to E2-dependent pathways. In rats, E4 has been shown to stimulate vasodilation of arteries via ERα activation [53, 54]. Studies of transgenic mice with mutations in the transactivation function (AF), AF1 or AF2 domain, have revealed that E4 induces atheroprotective effects through the activation of the genomic pathway of ERα [41]. Moreover, E4 exerted a protective effect against neointimal hyperplasia [55], a phenomenon described as being genomic-ERα dependent [56]. E4 was also shown to reduce the effects of angiotensin II and to prevent hypertension in rodents through the genomic-ERα pathway [55]. As predicted by the binding affinity constant (Table 2), the potency of E4 required to activate the genomic-ERα pathway is 50–100 times lower than that of E2. In contrast to E2, E4 did not accelerate endothelial healing in vivo or enhance endothelial nitric oxide synthase (eNOS) activation ex vivo [41, 55]; both of which are actions that have been established to be dependent on the MISS pathway of ERα [44, 57]. In addition, E4 inhibited the action of E2 on endothelial healing and eNOS activation, suggesting E4 antagonizes the E2-induced MISS pathway of ERα [41]. These results support that in the endothelium, E4 is an agonist of the genomic-ERα pathway but an antagonist of the MISS pathway (Fig. 3).

Estrogen signaling pathways induced by E4-only treatment or a combination of E2 + E4
E4 also acts through ERα to promote proliferation and growth of the mouse mammary gland [58], human ER + breast cancer cells [59, 60], patient-derived xenografts (PDX) from ER + breast tumors [60], and endometrial cancer cells [61]. However, in contrast to what was observed in the endothelium, E4 is reported to activate both the genomic-ERα and MISS pathways in breast cancer cells (Fig. 3) [41, 59]. In vitro, E4 stimulated rapid phosphorylation of serine 118 (S118)-ERα, the major phosphorylation site for activation of the receptor by the estrogens [59, 60]. When bound to ERα, E4 promotes the recruitment of the same set of co-regulators as E2, including PELP1 (proline-, glutamic acid-, and leucine-rich protein 1), MED1 (mediator complex subunit 1), SRC1 (steroid receptor coactivator 1), SRC2, SRC3, CBP/p300 (cAMP response element-binding protein), BRD8 (bromodomain-containing protein 8), LCoR (ligand-dependent corepressor), and NRIP1/RIP140 (nuclear receptor-interacting protein 1) [60]. Large scale RNAseq analyses performed on human MCF7 breast cancer cells revealed that, after 24 h incubation, E4 resulted in a similar transcriptomic profile with 97% of genes showing no differential expression in comparison to E2. E4 induced the expression and synthesis of genomic-related targets, such as the progesterone receptor (PR) [59, 60]. In MCF7 cells, E4 activated the MISS signaling pathway, by promoting the interaction between ERα and the tyrosine kinase Src, one of the first step in the MISS pathway [59]. It also induced rapid intracellular signaling cascades, including the MAPK/ERK1/2 and PI3K/AKT pathways. Interestingly, E4 in combination with E2 did not antagonize the E2-induced genomic-ERα pathway when assessed in vitro [41, 59]. Indeed, when E4 was combined with E2, it did not block the binding of ERα to ERE, or the transcription or synthesis of PR. However, E4 resulted in inhibition of the E2-induced MISS pathway in MCF7 cells, as it prevented the interaction between ERα and Src induced by E2 [41]. In breast cancer cells, E4 is an agonist of both the genomic-ERα and the MISS pathway. However, it can also antagonize the E2-induced MISS pathway.
In contrast to endocrine-sensitive ER + breast cancer cells, in endocrine-resistant ER + breast cancer cells E4 induces a distinct ERα-mediated signaling pathway. This pathway results in a gene expression signature associated with the unfolded protein response (UPR) and apoptosis; including downregulation of UPR genes associated with lipid metabolism (MBTPS1), endoplasmic reticulum-associated degradation (HTRA4, SYVN1, HERPUD1), and chaperone proteins involved in pro-survival mechanisms (e.g. SIL1) [61], and upregulation of CEBPB and INHBE, which are associated with high UPR stress. In endocrine-resistant ER + breast cancer cells, this gene signature is associated with a pro-apoptotic profile.
In summary, in all tissues evaluated so far, E4 is an agonist of the genomic-ERα pathway. However, activation of the MISS pathway appears to be tissue dependent. In the endothelium, E4 is an antagonist of the MISS pathway but when used alone in breast cancer cells it is an agonist of the MISS pathway. Interestingly, when combined with E2, E4 prevents activation of the MISS pathway induced by E2 in both the endothelium and in human endocrine-sensitive ER + breast cancer cells (Fig. 3).
Estetrol and Mammary Gland
To the best of our knowledge, there is only one study that has explored the effect of E4 on the mammary gland in prepubescent mice. It showed that E4 promoted elongation and proliferation of the murine mammary ductal tree in vivo by inducing the growth of prepubertal epithelial ducts and the appearance of terminal end buds (TEBs) [58]. Oral administration of E4 at a dose of between 0.3 to 10 mg/kg/day increased the growth of the murine mammary gland, but to a lesser extent than E2. Of note, the levels of E4 detected in the blood were 12–374 times higher than those of E2. Thus, in the murine mammary gland, E4 is considered as a weak estrogen in comparison to E2. This observation was also reported in human breast epithelial (HBE) cells in vitro, where E4 was 100 times less potent at stimulating proliferation of these cells than E2. Interestingly, when HBE cells and mice were treated with a combination of E2 and E4, E4 partially antagonized the strong stimulatory effect of E2 on HBE cell proliferation and murine mammary duct growth [58]. ERα is the receptor that mediates the estrogenic action of E4 on the mammary gland however, the mechanisms of action sustaining the anti-estrogenic effect have not yet been elucidated. Although these observations support the use of E4-based COCs for the prevention of E2-induced mammary gland proliferation in premenopausal women, the combination of E4 and a progestogen has not yet been evaluated in relation to mammary gland biology.
Estetrol and Breast Cancer
The effect of E4 on tumorigenesis in the breast and on breast cancer progression is complex and remains debated in the literature. Although E4 has primarily been described as pro-apoptotic, it has also been shown to have pro-tumoral effects on breast cancer [59, 62,63,64]. However, it is important to note the sensitivity status of the breast cancer cells and/or tumors studied with respect to endocrine therapy when interpreting such studies. Visser et al. [62], showed an anti-tumoral effect of E4 in a 7,12-dimethylben[a]antracene (DMBA)-induced breast cancer model in rat. In this study, daily oral administration of E4 prevented the initiation of breast tumors in a dose-dependent manner (2.5–10 mg/kg/day) in intact rats with endogenous levels of E2. At high doses (10 mg/kg/day), E4 suppressed breast tumor progression to a similar extent as ovariectomy and more efficiently than tamoxifen treatment [62]. In in vitro experiments performed on a long-term estrogen-deprived (LTED) human MCF7 breast cancer cell line [63] and in endocrine-resistant ER + breast cancer cells [61] E4 had pro-apoptotic properties similar to E2. In endocrine-resistant ER + breast cancer cells, these pro-apoptotic properties were associated with an ERα-dependent UPR [61]. Singer et al. [64], reported that when E4 was administrated orally (20 mg/day) for 2 weeks in women with recently diagnosed breast cancer, immunohistochemical assessment of tumors revealed an increased number of apoptotic cells but no modulation of the proliferative marker Ki67, when compared to placebo. This suggests that although E4 induced apoptosis in cancer cells, it does not have an impact on proliferation. In a multicenter, open-label, phase Ib/IIa, dose-escalation (20 mg, 40 mg, and 60 mg E4) study in postmenopausal women with resistant advanced breast cancer who have failed on anti-estrogen treatment with tamoxifen and aromatase inhibitors, E4 had an anti-tumor effect in five of nine patients. However, the three of three patients who received 60 mg E4 exhibited progressive tumor growth [65].
E4 had dose-dependent estrogenic properties, inducing pro-tumoral effects at high doses in human endocrine-sensitive breast cancer cells in vitro and in several breast tumor models in vivo [59, 60, 66]. E4 (10−8 M-10−5 M) increased the proliferation of MCF7 and T47D human ER + breast cancer cells, but with 50–100 less potency than E2. In addition to these proliferative effects, E4 reduced apoptosis in endocrine-sensitive breast cancer cells and reduced cell cytotoxicity [59]. E4 (10−9 M-10−7 M) also controlled ER + breast cancer cell (T47D) migration and invasion in a dose-dependent manner, through remodeling of the actin cytoskeleton via phosphorylation of moesin (Thr558); however, the pro-migratory effect of E4 was much lower than that of E2 [67]. In vivo, oral administration of E4 (3–10 mg/kg/day) increased the growth of MCF7 cells engrafted in ovariectomized immunodeficient mice in a dose-dependent manner, although a dose of 0.5 mgE4/kg/day was neutral [59]. These results were corroborated by work in MMTV-PyMT transgenic mice and PDXs from ER + breast cancer [60], which modelled hormonal human treatments by administering steroids to mice in a pattern that closely mimics daily oral steroid exposure in women. In contrast to E2 treatment, experiments using PDXs revealed that long term treatment with E4, delivered continuously over 30 weeks by Alzet pump (0.3 mg/kg/day) to mimic the therapeutic dose used for contraceptive purposes or in the treatment of symptoms due to menopause (15 mg/day), did not increase breast cancer growth. In addition, at this dose E4 treatment did not promote breast cancer metastasis dissemination to the lung in transgenic MMTV-PyMT mice [60]. However, at high pharmacological doses (3 mg/kg/day) exceeding 10 times the therapeutic dose of E4 reported to be required to treat symptoms due to menopause, E4 was pro-tumoral and pro-metastatic. This supratherapeutic dose of E4 (3 mg/kg/day) produces effects similar to the therapeutic dose of E2 required to treat symptoms due to menopause [60]. Interestingly, combined administration of E4 and E2 showed that E4 partially antagonized E2-induced breast cancer cell proliferation and migration in vitro (MCF7, T47D), and MCF7 breast tumor growth in vivo, when used at high doses of 10–8-10−6 M and 3–10 mg/kg/day, respectively [59, 67, 68]. These observations by Gérard et al. [59] in human MCF7 tumor xenografts and those of Visser et al. [62] in rat DMBA-induced breast cancer model, highlight the anti-estrogenic action of E4 on the pro-tumoral effect of exogenous and endogenous E2. This anti-estrogenic effect is not related to the antagonistic action of E4 on the genomic-ERα pathway. However, the actual molecular mechanisms employed to sustain this anti-estrogenic effect in endocrine-sensitive breast cancer cells are not yet fully understood.
E4 was also evaluated in combination with progesterone or drospirenone (DRSP) using three complementary endocrine-sensitive breast cancer models [60]: transgenic MMTV-PyMT mice, human MCF7 cell xenografts, and ER + breast cancer PDXs. Progesterone and DRSP were administered to mice in order to match human therapeutic doses. The combination of progesterone or DRSP and E4 (0.3 mg/kg/day) for use as a MHT did not impact tumor growth or metastasis dissemination when compared to E4 used alone. Moreover, the addition of progesterone did not potentiate the pro-tumoral effect observed at a supratherapeutic dose of E4 (3 mg/kg/day). Transcriptomics in MCF7 cells also showed that the addition of progesterone or its analog R5020 to E4 had a limited impact on gene transcription since only few genes were modulated. Nevertheless, several of the genes that were upregulated including, SGK1, FAM105A, FGF18, and TMEM63C, belong to a gene signature associated with progesterone-induced anti-proliferative effects related to good clinical outcomes [69]. These observations show that combining a therapeutic dose of E4 with progesterone or DRSP has a neutral impact on endocrine-sensitive breast cancer models in vivo.
Together, these data highlight the complex nature of E4 action in breast cancer. While the estrogenic actions of E4 promote pro-apoptotic features in endocrine-resistant breast cancer, in endocrine-sensitive breast cancers, E4 has a neutral impact even when combined with a progesterone or DRSP at the therapeutic dose for contraception or MHT. However, at doses exceeding this it becomes pro-tumoral in a dose-dependent manner. Notably, when combined with endogenous or exogenous E2, E4 can prevent the pro-tumoral action of E2. The effect of E4 on breast cancer cells is therefore highly dependent on the concentration or dose used and on whether or not it is combined with other estrogens, in particular E2, providing the opportunity to define a safe therapeutic window for use in different clinical applications.
Estetrol Potential for Therapeutic Indications
Preclinical and clinical studies have identified interesting therapeutic indications for E4, such as contraception, the treatment of symptoms due to menopause, and the treatment of endocrine-resistant breast cancer.
Contraception
Dose dependent inhibition of ovulation induced by E4 in cycling rats [70,71,72] was confirmed in a pilot phase II dose-finding study at a dose of 10 mg E4/day; with maximal inhibition seen at 20 mg E4/day [73]. Moreover, E4 treatment was also shown to decrease ovarian (E2) and gonadotropins (LH and FSH) hormone production [73]. The randomized phase II FIESTA study reported a favorable vaginal bleeding pattern and good cycle control associated with 15 mg E4/3 mg DRSP [74]. In addition, E4/DRSP treatment was also shown to be associated with favorable body weight control, well-being, high acceptability, and user satisfaction [75]. Since the combination of E4 and DRSP has been shown to have a neutral impact on breast cancer growth in vivo compared to E2 and progesterone [60], the use of an E4-based COC formulation could be advantageous in women who are concerned about the potential risk of breast cancer associated with conventional COCs. More interestingly, E4 did not increase the synthesis of sex hormone binging globulin (SHBG) [76,77,78] and had a limited effect on liver factors involved in coagulation, supporting a lower risk of cardiovascular side effects in comparison to E2 [55, 79]. This feature of E4 is possibly the most important advantage of E4-based formulations over conventional COCs.
Menopause Symptoms
Menopause is associated with the appearance of menopause symptoms, such as vasomotor symptoms (hot flushes and/or night sweats), genitourinary symptoms (vaginal dryness and atrophy, uterine bleeding and sexual dysfunction), urinary symptoms (incontinence and infections), mood change (irritability, anxiety, sadness, hyper-sensibility and depression), and cognitive disturbance [80, 81]. In addition, menopause is also associated with an increased risk of cardiovascular diseases, bone fractures related to osteoporosis, and metabolic complications such as type-2 diabetes [82,83,84,85]. Several preclinical and clinical studies have evaluated the impact of E4 on estrogen-sensitive tissues and organs related to menopause symptoms.
In an in vivo model of hot flushes in rats, E4 was shown to suppress vasomotor symptoms in a dose dependent manner [86]. E4 induced the synthesis of allopregnanolone in the brain of castrated rats, which is associated with body temperature regulation [87]. E4 also prevented E2-induced production of this molecule, highlighting the estrogenic and anti-estrogenic properties of E4 [88, 89]. Two clinical studies demonstrated that E4 (at a dose of 2–10 mg/day or 15 mg/day) effectively reduced the frequency and the severity of hot flushes in postmenopausal women [90, 91]. These results support a beneficial effect for E4 in controlling hot flushes.
In rodents, E4 promoted the proliferation of vaginal cells, increased vaginal weight and epithelium height [92], and induced vaginal cornification and maturation [93]. A randomized clinical study evaluating oral administration of E4 (at a dose of 2–40 mg/day) in postmenopausal women also revealed modifications to vaginal cytology, including a decrease in the percentage of parabasal cells and an increase in the quantity of superficial cells [90]. These preclinical and clinical studies show that E4 is associated with protective actions on the vagina and highlight a potential role for E4 in the prevention of vulvo-vaginal atrophy.
Interestingly, E4 attenuated brain injury in a neonatal rat model of hypoxic-ischemic encephalopathy, prevented oxidative stress and enhanced cell proliferation in primary hippocampal neuronal cell cultures in vitro, decreased early grey matter loss, and promoted neurogenesis and angiogenesis in vivo [94]. These results show that E4 presents neuroprotective properties and support that E4 could be investigated for the prevention of menopause symptoms related to cognitive function.
In another preclinical study, oral administration of E4 decreased levels of osteocalcin and increased bone density, mineral content, and bone strength in a dose dependent manner in ovariectomized rats [37]. In postmenopausal women, E4 also exerted a dose-dependent decrease in C-telopeptide and osteocalcine, markers of bone resorption and formation, respectively [70, 95]. At higher doses (20 and 40 mg), E4 stimulated bone formation, highlighting the potential use of E4 in the prevention of osteoporosis [70]. These results highlight promising clinical benefits and a potential role for E4 on bone fractures and osteoporosis risk.
Additional positive effects of E4 were also reported on menopause-associated risks, including metabolic disorders and cardiovascular issues. In mouse models, E4 reduced body weight gain, improved glucose tolerance, prevented obesity [96], and associated disorders such as atherosclerosis and steatosis [53]. Preclinical and clinical studies revealed that E4 has several potential vascular advantages (reviewed in [55]), including the prevention of angiotensin-II-dependent hypertension and neointimal hyperplasia, while having minimal impact on hemostasis, fibrinolysis, angiotensinogen, triglycerides, and cholesterol. Although E4 did not enhance eNOS action in murine adult aorta [41, 55], it favored flow-induced vasodilation [53, 54], an important vasculoprotective action of estrogen. In addition, E4 also prevented atherosclerosis in a dose-dependent manner in mice [41].
Importantly, preclinical and clinical studies demonstrated that E4 induces uterotrophic activity and acts as an agonist on the endometrium [41, 90]. It has also been shown to increase mouse uterine wet weight [37, 93, 97], epithelial proliferation, and height of the epithelium and stromal compartments [41] at a MHT therapeutic dose [60]. In rats, E4 increased the volume of luminal fluids, protein content, and the activity of alkaline phosphatase [97]; all markers of the estrogenic uterine response. Moreover, it induced major histological modifications, including the synthesis of the PR [98]. In postmenopausal women, E4 (10 mg/day) increased the thickness of the endometrium, highlighting its estrogenic action [90]. The addition of DRSP to E4 (5 or 10 mg/day) decreased E4-induced endometrial thickening in women [73]. These results support the addition of a progestogen to E4 formulations for MHT to protect the endometrium of non-hysterectomized women from hyperplasia and cancer.
Assessing the risk of breast cancer associated with E4 use in postmenopausal women is a long-term effort and can only be conducted with decades of patient follow-up. Nevertheless, as detailed in the previous section of this review, preclinical data obtained from several endocrine-sensitive breast cancer models, including PDX, revealed that E4 has a neutral impact on breast cancer growth and metastasis dissemination to the lung when used in mice at a dose (0.3 mg/kg/day) that corresponds to the steady-state obtained by once-a-day (15 mg E4/day) oral treatment in women. This neutral effect is not impacted by the addition of progesterone or DRSP [60]. These observations give E4-based formulations an advantage over E2-based MHTs and have important clinical implications as they highlight the possibility of developing a combined estrogen, progestogen MHT that could have a positive impact on breast cancer risk.
Breast Cancer Treatment
Acquired endocrine resistance is a major cause of relapse in ER + breast cancer and therapeutic strategies to help overcome this are of the utmost importance. Several studies have suggested the potential of estrogen-based therapies in the treatment of advanced endocrine-resistant breast cancer. However, there is a reluctance to use E2 due to the potential for adverse effects, especially thromboembolism [99,100,101,102,103,104]. The pro-apoptotic properties of E4 on endocrine-resistant breast cancer demonstrated in vitro [61] and in clinical trials [64, 65], in addition to the limited effect of E4 on liver factors involved in coagulation [79], support a potential role for E4 in the treatment of advanced endocrine-resistant breast cancer in postmenopausal women.
Conclusions and Perspectives
Of the natural estrogens, E4 is a unique native fetal estrogen with selective tissue actions that offers novel therapeutic opportunities for indications including, contraception, the treatment of symptoms due to menopause, and the treatment of endocrine-resistant advanced breast cancer.
The safety data for E4 in the breast are promising, nevertheless further progress in this field is expected, especially regarding the mechanisms of action of this natural estrogen on the mammary gland and breast cancer. A recent publication emphasized that, in addition to the genomic-ERα pathway [105], the MISS-ERα pathway also plays a role in promoting intercellular communication during mammary gland development [106]. Since E4 is characterized as an antagonist and an agonist of the MISS pathway depending on the tissue, it is important to fully characterize the molecular impact of E4 on mammary gland biology. In breast cancer, preclinical and clinical studies have shown that E4 formulations have both pro-tumoral and pro-apoptotic effects depending on the dose and whether or not it is combined with endogenous or exogenous E2. The molecular mechanisms leading to the anti-estrogenic action of E4 in particular, remain to be fully elucidated.
In conclusion, E4 does not meet every characteristic of the ideal estrogen. However, in comparison to other conventional estrogens, E4 does meet several important criteria. E4 could be considered as a friend of the mammary gland, even when combined to progesterone or DRSP to protect the endometrium of non-hysterectomized women, since it remains neutral on preclinical models of breast cancer at a dose that is effective at preventing hot flushes; a symptom of menopause that arises in 80% of postmenopausal women. However, at high dose, E4 remains a foe of the mammary gland highlighting the importance of exerting extreme caution when determining the dose required for management versus prevention of mammary side effects. E4 also displays cardioprotective features against atherosclerosis and has a limited impact on liver factors involved in coagulation, supporting a lower risk of thromboembolic events and thromboembolism. These features support a safer profile in terms of breast cancer risk and thromboembolism risk making E4 a safer estrogenic treatment option for women.
References
Vasquez YM. Estrogen-regulated transcription: Mammary gland and uterus. Steroids. 2018;133:82–6.
Hamilton KJ, Hewitt SC, Arao Y, Korach KS. Estrogen hormone biology. Curr Top Dev Biol. 2017;125:109–46.
Brisken C, Ataca D. Endocrine hormones and local signals during the development of the mouse mammary gland. Wiley Interdiscip Rev Dev Biol. 2015;4:181–95.
Baudry M, Bi X, Aguirre C. Progesterone-estrogen interactions in synaptic plasticity and neuroprotection. Neuroscience. 2013;239:280–94.
Krassas GE, Papadopoulou P. Oestrogen action on bone cells. J Musculoskel Neuron Interact. 2001;2:143–51.
Meyer MR, Clegg DJ, Prossnitz ER, Barton M. Obesity, insulin resistance and diabetes: sex differences and role of oestrogen receptors. Acta Physiol. 2011;203:259–69.
Handgraaf S, Riant E, Fabre A, Waget A, Burcelin R, Lière P, et al. Prevention of obesity and insulin resistance by estrogens requires ERα activation function-2 (ERαAF-2), whereas ERαAF-1 is dispensable. Diabetes. 2013;62:4098–108.
Thornton MJ. Estrogens and aging skin. Dermatoendocrinol. 2013;5:264–70.
Toutain CE, Brouchet L, Raymond-Letron I, Vicendo P, Bergès H, Favre J, et al. Prevention of skin flap necrosis by estradiol involves reperfusion of a protected vascular network. Circ Res. 2009;104:245–54.
Ansar Ahmed S, Penhale WJ, Talal N. Sex hormones, immune responses, and autoimmune diseases. Mechanisms of sex hormone action. Am J Pathol. 1985;121:531–51.
Pavlidou E, et al. Menopause hormonal treatments in 2016. Med Rev from Switz. 2016;12:1805–10.
Pinkerton JAV, Aguirre FS, Blake J, Cosman F, Hodis H, Hoffstetter S, et al. The 2017 hormone therapy position statement of the North American Menopause Society. Menopause. 2017;24:728–53.
Tanos T, Rojo LJ, Echeverria P, Brisken C. ER and PR signaling nodes during mammary gland development. Breast Cancer Res. 2012;14:1–12.
Trémollieres F. Oral combined contraception: Is there any difference between ethinyl-estradiol and estradiol?. Gynecol Obstet Fertil. 2012;40:109–15.
Collaborative Group on Hormonal Factors in Breast. Breast cancer and hormonal contraceptives: collaborative reanalysis of individual data on 53 297 women with breast cancer and 100 239 women without breast cancer from 54 epidemiological studies. Lancet. 1996;347:1713–27.
Mørch LS, Skovlund CW, Hannaford PC, Iversen L, Fielding S, Lidegaard Ø. Contemporary hormonal contraception and the risk of breast cancer. N Engl J Med. 2017;377:2228–39.
Iversen L, Sivasubramaniam S, Lee AJ, Fielding S, Hannaford PC. Lifetime cancer risk and combined oral contraceptives: the Royal College of General Practitioners’ Oral Contraception Study. Am J Obstet Gynecol. 2017;216:580.e1-580.e9.
Hannaford PC, Selvaraj S, Elliott AM, Angus V, Iversen L, Lee AJ. Cancer risk among users of oral contraceptives: cohort data from the Royal College of General Practitioner’s oral contraception study. Br Med J. 2007;335:1–8.
Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results from the Women’s Health Initiative randomized controlled trial. J Am Med Assoc. 2002;288:321–33.
Wysowski DK, Governale LA. Use of menopausal hormones in the United States, 1992 through June, 2003. Pharmacoepidemiol Drug Saf. 2004;14:171–6.
Banks E, Beral V, Bull D, Reeves G, Austoker J, English R, et al. Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet Lancet Publishing Group. 2003;362:419–27.
Fournier A, Berrino F, Clavel-Chapelon F. Unequal risks for breast cancer associated with different hormone replacement therapies: Results from the E3N cohort study. Breast Cancer Res Treat. 2008;107:103–11.
Lyytinen H, Pukkala E, Ylikorkala O. Breast cancer risk in postmenopausal women using estradiol-progestogen therapy. Obstet Gynecol. 2009;113:65–73.
Bakken K, Fournier A, Lund E, Waaseth M, Dumeaux V, Clavel-Chapelon F, et al. Menopausal hormone therapy and breast cancer risk: Impact of different treatments. The European Prospective Investigation into Cancer and Nutrition. Int J Cancer. 2011;128:144–56.
Fournier A, Berrino F, Riboli E, Avenel V, Clavel-Chapelon F. Breast cancer risk in relation to different types of hormone replacement therapy in the E3N-EPIC cohort. Int J Cancer Int J Cancer. 2005;114:448–54.
Marjoribanks J, Farquhar C, Roberts H, Lethaby A. Long term hormone therapy for perimenopausal and postmenopausal women. Cochrane Database Syst Rev. 2012;7:CD004143.
Holm M, Olsen A, Au Yeung S, Overvad K, Lidegaard Ø, Kroman N, et al. Pattern of mortality after menopausal hormone therapy: long-term follow up in a population-based cohort. BJOG An Int J Obstet Gynaecol. 2019;126:55–63.
Beral V, Peto R, Pirie K. Type and timing of menopausal hormone therapy and breast cancer risk: individual participant meta-analysis of the worldwide epidemiological evidence. Lancet Lancet Publishing Group. 2019;394:1159–68.
Beral V, Bull D, Doll R, Key T, Peto R, Reeves G, et al. Breast cancer and hormone replacement therapy: collaborative reanalysis of data from 51 epidemiological studies of 52,705 women with breast cancer and 108,411 women without breast cancer. The Lancet. 1997;350:1047–59.
Hagen AA, Barr M, Diczfalusy E. Metabolism of 17-Beta-estradiol-4-14-C in early infancy. Acta Endocrinol (Copenh). 1965;49:207–20.
Cantineau R, Kremers P, De Graeve J, Gielen JE, Lambotte R. 15- and 16-hydroxylations of androgens and estrogens in the human fetal liver: A critical step in estetrol biosynthesis. J Steroid Biochem. 1985;22:195–201.
Holinka CF, Diczfalusy E, Coelingh Bennink HJT. Estetrol: A unique steroid in human pregnancy. Climacteric. 2008;11:1–10.
Coelingh Bennink F, Holinka CF, Visser M, Coelingh Bennink HJT. Maternal and fetal estetrol levels during pregnancy. Climacteric. 2008;11:69–72.
Tulchinsky D, Frigoletto FD, Ryan KJ, Fishman J. Plasma estetrol as an index of fetal well-being. J Clin Endocrinol Metab. 1975;40:560–7.
Holinka CF, Diczfalusy E, Coelingh Bennink HJT. Estetrol: A unique steroid in human pregnancy. J Steroid Biochem Mol Biol Pergamon. 2008;110:138–43.
Visser M, Holinka CF, Coelingh Bennink HJT. First human exposure to exogenous single-dose oral estetrol in early postmenopausal women. Climacteric. 2008;11:31–40.
Coelingh Bennink HJT, Heegaard AM, Visser M, Holinka CF, Christiansen C. Oral bioavailability and bone-sparing effects of estetrol in an osteoporosis model. Climacteric Taylor & Francis. 2008;11:2–14.
Visser M, Foidart JM, Coelingh Bennink HJT. In vitro effects of estetrol on receptor binding, drug targets and human liver cell metabolism. Climacteric. 2008;11:64–8.
Fishman J. Fate of 15α-hydroxyestriol-3H in adult man. J Clin Endocrinol Metab. 1970;31:436–8.
Jirku H, Kadner S, Levitz M. Pattern of estetrol conjugation in the human. Steroids Elsevier. 1972;19:519–34.
Abot A, Fontaine C, Buscato M, Solinhac R, Flouriot G, Fabre A, et al. The uterine and vascular actions of estetrol delineate a distinctive profile of estrogen receptor α modulation, uncoupling nuclear and membrane activation. EMBO Mol Med. 2014;6:1328–46.
Billon-Galés A, Fontaine C, Filipe C, Douin-Echinard V, Fouque MJ, Flouriot G, et al. The transactivating function 1 of estrogen receptor α is dispensable for the vasculoprotective actions of 17 β-estradiol. Proc Natl Acad Sci. 2009;106:2053–8.
Billon-Galés A, Krust A, Fontaine C, Abot A, Flouriot G, Toutain C, et al. Activation function 2 (AF2) of estrogen receptor-α is required for the atheroprotective action of estradiol but not to accelerate endothelial healing. Proc Natl Acad Sci U S A. 2011;108:13311–6.
Adlanmerini M, Solinhac R, Abot A, Fabre A, Raymond-Letron I, Guihot AL, et al. Mutation of the palmitoylation site of estrogen receptor a in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc Natl Acad Sci U S A. 2014;111:E283–90.
Abot A, Fontaine C, Raymond-Letron I, Flouriot G, Adlanmerini M, Buscato M, et al. The AF-1 activation function of estrogen receptor α is necessary and sufficient for uterine epithelial cell proliferation in vivo. Endocrinology Oxford Academic. 2013;154:2222–33.
Arao Y, Hamilton KJ, Ray MK, Scott G, Mishina Y, Korach KS. Estrogen receptor α AF-2 mutation results in antagonist reversal and reveals tissue selective function of estrogen receptor modulators. Proc Natl Acad Sci U S A. 2011;108:14986–91.
Brouchet L, Krust A, Dupont S, Chambon P, Bayard F, Arnal JF. Estradiol accelerates reendothelialization in mouse carotid artery through estrogen receptor-alpha but not estrogen receptor-beta. Circulation. 2001;103:423–8.
Khbouz B, Bournonville C, Court L, Taziaux M, Corona R, Arnal J, et al. Role for the membrane estrogen receptor alpha in the sexual differentiation of the brain. Eur J Neurosci. 2020;52:2627–45.
Pedram A, Razandi M, Lewis M, Hammes S, Levin ER. Membrane-localized estrogen receptor α is required for normal organ development and function. Dev Cell Cell Press. 2014;29:482–90.
Arnal J-F, Lenfant F, Metivier R, Flouriot G, Henrion D, Adlanmerini M, et al. Membrane and nuclear estrogen receptor alpha actions: From tissue specificity to medical implications. Physiol Rev. 2017;97:1045–87.
Arnal JF, Fontaine C, Abot A, Valera MC, Laurell H, Gourdy P, et al. Lessons from the dissection of the activation functions (AF-1 and AF-2) of the estrogen receptor alpha in vivo. Steroids Elsevier. 2013;78:576–82.
Soltysik K, Czekaj P. Membrane estrogen receptors - is it an alternative way of estrogen action?. J Physiol Pharmacol. 2013;64:129–42.
Hilgers RHP, Oparil S, Wouters W, Coelingh Bennink HJT. Vasorelaxing effects of estetrol in rat arteries. J Endocrinol. 2012;215:97–106.
Levine MG, Miodovnik M, Clark KE. Uterine vascular effects of estetrol in nonpregnant ewes. Am J Obstet Gynecol. 1984;148:735–8.
Foidart J, Gaspard U, Péqueux C, Jost M, Gordenne V, Tskitishvili E, et al. Unique vascular benefits of estetrol, a native fetal estrogen with specific actions in tissues (NEST). In: ISGE Series; Brinton R.D., Genazzani A.R., Simoncini T. et al., Sex steroids. Effects on brain, heart and vessels. Int Soc Gynecol Endocrinol. 2019. pp. 169–95.
Pedram A, Razandi M, Kim JK, O’Mahony F, Lee EYHP, Lederer U, et al. Developmental phenotype of a membrane only estrogen receptor α (MOER) mouse. J Biol Chem. 2009;284:3488–95.
Chambliss KL, Wu Q, Oltmann S, Konaniah ES, Umetani M, Korach KS, et al. Non-nuclear estrogen receptor α signaling promotes cardiovascular protection but not uterine or breast cancer growth in mice. J Clin Invest. 2010;120:2319–30.
Gérard C, Blacher S, Communal L, Courtin A, Tskitishvili E, Mestdagt M, et al. Estetrol is a weak estrogen antagonizing estradiol-dependent mammary gland proliferation. J Endocrinol BioScientifica Ltd. 2014;224:85–95.
Gérard C, Mestdagt M, Tskitishvili E, Communal L, Gompel A, Silva E, et al. Combined estrogenic and anti-estrogenic properties of estetrol on breast cancer may provide a safe therapeutic window for the treatment of menopausal symptoms. Oncotarget. 2015;6:17621–36.
Gallez A, Blacher S, Maquoi E, Konradowski E, Joiret M, Primac I, et al. Estetrol combined to progestogen for menopause or contraception indication is neutral on breast cancer. Cancers. 2021;13(2486):1–23.
Abderrahman B, Maximov PY, Curpan RF, Hanspal JS, Fan P, Xiong R, Tonetti DA, Thatcher GRJ, Jordan VC. Pharmacology and molecular mechanisms of clinically relevant estrogen estetrol and estrogen mimic BMI-135 for the treatment of endocrine-resistant breast cancer. Mol Pharmacol. 2020;98:364–81.
Visser M, Kloosterboer HJ, Coelingh Bennink HJT. Estetrol prevents and suppresses mammary tumors induced by DMBA in a rat model. Horm Mol Biol Clin Investig. 2012;9:95–103.
Yue W, Verhoeven C, Bernnink HC, Wang JP, Santen RJ. Pro-apoptotic effects of estetrol on long-term estrogen-deprived breast cancer cells and at low doses on hormone-sensitive cells. Breast Cancer Basic Clin Res. 2019;13:1–8.
Singer CF, Bennink HJTC, Natter C, Steurer S, Rudas M, Moinfar F, et al. Antiestrogenic effects of the fetal estrogen estetrol in women with estrogen-receptor positive early breast cancer. Carcinogenesis. 2014;35:2447–51.
Schmidt M, Lenhard H, Hoenig A, Zimmerman Y, Krijgh J, Jansen M, Coelingh Bennink HJT. Tumor suppression, dose-limiting toxicity and wellbeing with the fetal estrogen estetrol in patients with advanced breast cancer. J Cancer Res Clin Oncol. 2021;147:1833–42.
Jozan S, Kreitmann B, Bayard F. Different effects of oestradiol, oestriol, oestetrol and of oestrone on human breast cancer cells (MCF-7) in long term tissue culture. Acta Endocrinol. 1981;98:73–80.
Giretti MS, Guevara MMM, Cecchi E, Mannella P, Palla G, Spina S, et al. Effects of estetrol on migration and invasion in T47-D breast cancer cells through the actin cytoskeleton. Front Endocrinol Frontiers Research Foundation. 2014;5:1–8.
Lippert C, Seeger H, Mueck AO. The effect of endogenous estradiol metabolites on the proliferation of human breast cancer cells. Life Sci. 2003;72:877–83.
Mohammed H, Russell IA, Stark R, Rueda OM, Hickey TE, Tarulli GA, et al. Progesterone receptor modulates ERα action in breast cancer. Nature Nature Publishing Group. 2015;523:313–7.
Visser M, Coelingh Bennink HJT. Clinical applications for estetrol. J Steroid Biochem Mol Biol Pergamon. 2009;114:85–9.
Coelingh Bennink HJT, Holinka CF, Diczfalusy E. Estetrol review: profile and potential clinical applications. Climacteric. 2008;11:47–58.
Coelingh Bennink HJT, Skouby S, Bouchard P, Holinka CF. Ovulation inhibition by estetrol in an in vivo model. Contraception. 2008;77:186–90.
Duijkers IJM, Klipping C, Zimmerman Y, Appels N, Jost M, Maillard C, et al. Inhibition of ovulation by administration of estetrol in combination with drospirenone or levonorgestrel: Results of a phase II dose-finding pilot study. Eur J Contracept Reprod Heal Care. 2015;20:476–89.
Apter D, Zimmerman Y, Beekman L, Mawet M, Maillard C, Foidart JM, et al. Bleeding pattern and cycle control with estetrol-containing combined oral contraceptives: Results from a phase II, randomised, dose-finding study (FIESTA). Contraception. 2016;94:366–73.
Apter D, Zimmerman Y, Beekman L, Mawet M, Maillard C, Foidart J-M, et al. Estetrol combined with drospirenone: an oral contraceptive with high acceptability, user satisfaction, well-being and favourable body weight control. Eur J Contracept Reprod Heal Care. 2017;22:260–7.
Mawet M, Maillard C, Klipping C, Zimmerman Y, Foidart JM, Bennink HJTC. Unique effects on hepatic function, lipid metabolism, bone and growth endocrine parameters of estetrol in combined oral contraceptives. Eur J Contracept Reprod Heal Care. 2015;20:463–75.
Hammond GL, Hogeveen KN, Visser M, Coelingh Bennink HJT. Estetrol does not bind sex hormone binding globulin or increase its production by human HepG2 cells. Climacteric. 2008;11:41–6.
Valéra M-C, Noirrit-Esclassan E, Dupuis M, Fontaine C, Lenfant F, Briaux A, et al. Effect of estetrol, a selective nuclear estrogen receptor modulator, in mouse models of arterial and venous thrombosis. Mol Cell Endocrinol. 2018;477:132–9.
Kluft C, Zimmerman Y, Mawet M, Klipping C, Duijkers IJM, Neuteboom J, et al. Reduced hemostatic effects with drospirenone-based oral contraceptives containing estetrol vs. ethinyl estradiol. Contraception. 2017;95:140–7.
Broekmans FJ, Soules MR, Fauser BC. Ovarian aging: Mechanisms and clinical consequences. Endocr Rev. 2009;30:465–93.
Mehta J, Kling JM, Manson JE. Risks, benefits, and treatment modalities of menopausal hormone therapy: Current concepts. Front Endocrinol. 2021;12:1–14.
Cui J, Shen Y, Li R. Estrogen synthesis and signaling pathways during aging: From periphery to brain. Trends Mol Med NIH Public Access. 2013;19:197–209.
Al-Safi ZA, Santoro N. Menopausal hormone therapy and menopausal symptoms. Fertil Steril. 2014;101:905–15.
Santoro N, Epperson CN, Mathews SB. Menopausal symptoms and their management. Endocrinol Metab Clin North Am. 2015;44:497–515.
Nelson H, Haney E, Humphrey L, Miller J, Nedrow A, Nicolaidis C, et al. Management of menopause-related symptoms: Summary. Agency Healthc Res Qual. 2005;120:1–10.
Holinka CF, Brincat M, Coelingh Bennink HJT. Preventive effect of oral estetrol in a menopausal hot flush model. Climacteric. 2008;11:15–21.
Ishiwata T, Saito T, Hasegawa H, Yazawa T, Kotani Y, Otokawa M, et al. Changes of body temperature and thermoregulatory responses of freely moving rats during GABAergic pharmacological stimulation to the preoptic area and anterior hypothalamus in several ambient temperatures. Brain Res. 2005;1048:32–40.
Pluchino N, Santoro AN, Casarosa E, Giannini A, Genazzani A, Russo M, et al. Effect of estetrol administration on brain and serum allopregnanolone in intact and ovariectomized rats. J Steroid Biochem Mol Biol. 2014;143:285–90.
Pluchino N, Drakopoulos P, Casarosa E, Freschi L, Petignat P, Yaron M, et al. Effect of estetrol on beta-endorphin level in female rats. Steroids. 2015;95:104–10.
Coelingh Bennink HJT, Verhoeven C, Zimmerman Y, Visser M, Foidart JM, Gemzell-Danielsson K. Clinical effects of the fetal estrogen estetrol in a multiple-rising-dose study in postmenopausal women. Maturitas. 2016;91:93–100.
Gaspard U, Taziaux M, Mawet M, Jost M, Gordenne V, Coelingh Bennink HJT, et al. A multicenter, randomized study to select the minimum effective dose of estetrol (E4) in postmenopausal women (E4Relief). Menopause. 2020;27:848–57.
Benoit T, Valera M-C, Fontaine C, Buscato M, Lenfant F, Raymond-Letron I, et al. Estetrol, a fetal selective estrogen receptor modulator, acts on the vagina of mice through nuclear estrogen receptor α activation. Am J Pathol. 2017;187:2499–507.
Heegaard AM, Holinka CF, Kenemans P, Coelingh Bennink HJT. Estrogenic uterovaginal effects of oral estetrol in the modified Allen-Doisy test. Climacteric. 2008;11:22–8.
Tskitishvili E, Nisolle M, Munaut C, Pequeux C, Gerard C, Noel A, et al. Estetrol attenuates neonatal hypoxic-ischemic brain injury. Exp Neurol. 2014;261:298–307.
Coelingh Bennink HJT, Verhoeven C, Zimmerman Y, Visser M, Foidart J-M, Gemzell-Danielsson K. Pharmacodynamic effects of the fetal estrogen estetrol in postmenopausal women: results from a multiple-rising-dose study. Menopause. 2017;24:677–85.
Buscato M, Davezac M, Zahreddine R, Adlanmerini M, Métivier R, Fillet M, Cobraiville G, Moro C, Foidart J-M, et al. Estetrol prevents western diet–induced obesity and atheroma independently of hepatic estrogen receptor α. Am J Physiol - Endocrinol Metab. 2021;320:E19-29.
Holinka CF, Gurpide E. In vivo effects of estetrol on the immature rat uterus. Biol Reprod Oxford Academic. 1979;20:242–6.
Holinka CF, Bressler RS, Zehr DR, Gurpide E. Comparison of effects of estetrol and tamoxifen with those of estriol and estradiol on the immature rat uterus. Biol Reprod. 1980;22:913–26.
Anderson GL, Limacher M. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: The women’s health initiative randomized controlled trial. J Am Med Assoc. 2004;291:1701–12.
Roehm E. A reappraisal of women’s health initiative estrogen-alone trial: Long-term outcomes in women 50–59 years of age. Obstet Gynecol Int. 2015;2015:1–10.
Chlebowski RT, Anderson GL, Aragaki AK, Manson JE, Stefanick ML, Pan K, et al. Association of menopausal hormone therapy with breast cancer incidence and mortality during long-term follow-up of the women’s health initiative randomized clinical trials. J Am Med Assoc. 2020;324:369–80.
Ellis MJ, Gao F, Dehdashti F, Jeffe DB, Marcom PK, Carey LA, et al. Lower-dose vs high-dose oral estradiol therapy of hormone receptor-positive, aromatase inhibitor-resistant advanced breast cancer: A phase 2 randomized study. J Am Med Assoc. 2009;302:774–80.
Iwase H, Yamamoto Y, Yamamoto-Ibusuki M, Murakami KI, Okumura Y, Tomita S, et al. Ethinylestradiol is beneficial for postmenopausal patients with heavily pre-treated metastatic breast cancer after prior aromatase inhibitor treatment: A prospective study. Br J Cancer. 2013;109:1537–42.
Lønning PE, Taylor PD, Anker G, Iddon J, Wie L, Jørgensen LM, et al. High-dose estrogen treatment in postmenopausal breast cancer patients heavily exposed to endocrine therapy. Breast Cancer Res Treat. 2001;67:111–6.
Cagnet S, Ataca D, Sflomos G, Aouad P, Schuepbach-Mallepell S, Hugues H, Krust A, et al. Oestrogen receptor α AF-1 and AF-2 domains have cell population-specific functions in the mammary epithelium. Nat Commun. 2018;9:4723:1–15.
Gagniac L, Rusidzé M, Boudou F, Cagnet S, Adlanmerini M, Jeannot P, Gaide N, Giton F, et al. Membrane expression of the estrogen receptor ERα is required for intercellular communications in the mammary epithelium. Development. 2020;147:1–15.
Funding
The authors are funded by the University of Liège, the Fondation Léon Fredericq, the Belgium National Fund for Scientific Research (FNRS) and the Télévie.
Author information
Authors and Affiliations
Contributions
A.G. wrote the original draft and reviewed the manuscript. I. DDS., V.W., and JM.F. critically reviewed the manuscript. C.P. acquired funding, supervised and critically reviewed the manuscript.
Corresponding author
Ethics declarations
Conflicts of Interest/Competing Interests
Jean-Michel Foidart is a member of the board of Mithra Pharmaceuticals (Belgium). Anne Gallez was a post-doctoral fellow at the University of Liège when developing this manuscript. She became an employee of Mithra Pharmaceuticals (Belgium) whilst this manuscript was under review by the journal. The other authors declared no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gallez, A., Dias Da Silva, I., Wuidar, V. et al. Estetrol and Mammary Gland: Friends or Foes?. J Mammary Gland Biol Neoplasia 26, 297–308 (2021). https://doi.org/10.1007/s10911-021-09497-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10911-021-09497-0