
Abstract
Puberty is a complex developmental process that varies considerably among individuals and populations. Genetic factors explain a large proportion of the variability of several pubertal traits. Recent genome-wide association studies (GWAS) have identified hundreds of variants involved in traits that result from body growth, like adult height. However, they do not capture many genetic loci involved in growth changes over distinct growth phases. Further, such GWAS have been mostly performed in Europeans, but it is unknown how these findings relate to other continental populations. In this study, we analyzed the genetic basis of three pubertal traits; namely, peak height velocity (PV), age at PV (APV) and height at APV (HAPV). We analyzed a cohort of 904 admixed Chilean children and adolescents with European and Mapuche Native American ancestries. Height was measured on roughly a \(6-\)month basis from childhood to adolescence between 2006 and 2019. We predict that, in average, HAPV is 4.3 cm higher in European than in Mapuche adolescents (P = 0.042), and APV is 0.73 years later in European compared with Mapuche adolescents (P = 0.023). Further, by performing a GWAS on 774, 433 single-nucleotide polymorphisms, we identified a genetic signal harboring 3 linked variants significantly associated with PV in boys (P \(< 5 \times 10^{-8}\)). This signal has never been associated with growth-related traits.
Similar content being viewed by others


Introduction
Puberty is a complex developmental process characterized by distinct as well as shared phenotypes in males and females. For instance, while in boys voice breaking represents a developmental milestone, in females distinct pubertal markers are menarche, namely the onset of the first menstrual bleed (Day et al. 2015a); and the onset of breast development. Among shared pubertal phenotypes in males and females are the onset of pubic hair as well as the pubertal growth spurt, also called peak height velocity (PV), which is the period where maximum rate of growth occurs (Biro et al. 2006). Importantly, deviations from normal pubertal growth correlate with adult risk for certain kinds of cancer, diabetes and cardiometabolic disorders (Cousminer et al. 2013; Day et al. 2015b).
Pubertal parameters are also affected by genetic ancestry. For example, the age at onset of puberty, defined as the age at specific areolar and pubic hair stages, is earlier in African American girls than in European American girls (9.6 vs. 10.2 years, respectively). This is also true for the age at PV (APV; 11.5 vs. 11.9 years), age at menarche (12.0 vs. 12.6 years) and the age at attainment of adult height (16.5 vs. 17.1 years) (Biro et al. 2006). Maternal obesity significantly affects the onset of pubic hair in Asian and non-Hispanic girls, but not in African American girls. Among boys, the onset of pubic hair development occurs earlier in African Americans than in European Americans (Euling et al. 2008). However, such studies present some drawbacks. (i) Ancestry is considered as that reported by the participants’ parents and is defined as a categorical variable, thus not accounting for different degrees of genetic admixture. (ii) Social and cultural disparities that correlate with phenotypic differences between ethnic groups are usually not rigorously accounted for (Idossa et al. 2018). (iii) Most studies have analyzed populations with African and European ancestries. Hence, it is not known whether or not—and to which extent—the observed results hold true for other continental groups, such as populations with Native American ancestry.
The onset of puberty is triggered by a combination of genetic and environmental factors. Environmental factors account for \(\sim 20-30\%\) of the variance of puberty’s onset and include nutrition, stress, life setting (urban vs. rural) and social status (Soliman et al. 2014; Herbison 2007; Parent et al. 2003). However, the major determinants of pubertal growth variability are genetic, since its heritability is 60–90% (Cousminer et al. 2013).
Genome-wide association studies (GWAS) have been key in identifying genetic variation involved in traits that result from body growth, such as adult height, which is highly heritable and polygenic, involving \(>3,000\) genetic variants (Chan et al. 2015; Styrkarsdottir et al. 2019). However, it is mostly unknown how these variants influence height growth across distinct growth phases and populations (Paternoster et al. 2011; Mills and Rahal 2020). A few GWA studies performed on European populations have identified loci involved in height growth traits. For example, a GWA meta-analysis performed on a pooled set of European cohorts identified loci involved in the take-off phase of the growth spurt (height at 10 years in girls and 12 years in boys), the total magnitude of height growth during the pubertal growth spurt, the timing of puberty and age at menarche (Elks et al. 2010; Cousminer et al. 2013). Nevertheless, with the exception of age at menarche (Cousminer et al. 2016), non-European populations have been poorly analyzed in such studies, making difficult to estimate the genetic contribution of associated loci in populations with other continental ancestries.
The Chilean population consists of admixed individuals whose continental ancestry comes from Europe (\(52\%\) on average), Native America (\(45\%\) on average) and Africa (\(3\%\) on average) (Eyheramendy et al. 2015). Their Native American ancestry component is made up of two sub-components, mainly Mapuche (from the lowlands of Central-Southern Chile) and Aymara (from the Andes highlands of Northern Chile) to a lower extent (Vicuña et al. 2020). While the general aim of this study was to identify genetic factors involved in pubertal height growth, our specific aims were twofold: (i) identifying novel genetic loci associated with PV, APV and HAPV; (ii) estimating the contribution of genetic ancestry in the variability of these traits. We implemented a GWAS in an Chilean cohort of 904 admixed individuals with European and Mapuche Native American ancestry, for whom these traits were measured from childhood to adolescence on a 6-month basis between 2010 and 2019.
Results
Characterization of PV, APV and HAPV in Chilean children
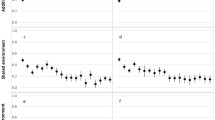
We modeled pubertal height growth in 440 Chilean boys and 464 girls separately. Figure 1 shows the growth curves for boys and girls (left and right panels in the top row, respectively) as well as the mean height velocity (left and right panels in the lower row, respectively). From the models, we estimated that the mean PV is 9.1 (±0.9 SD) cm/year in boys and 7.7 (±0.8 SD) cm/year in girls. The mean APV is 12.7 (±0.8 SD) years in boys and 10.8 (±0.8 SD) years in girls. We also found that HAPV is 156.4 (±5.5 SD) cm in boys and 145.1 (±5.4 SD) cm in girls (upper row).

Estimation of PV and APV. The upper row shows the adjustment of the growth curves in boys (left) and girls (right), obtained using the SITAR package from R (Cao et al. 2018). The lower row shows the mean height velocity (cm/year) in boys (left) and girls (right). The dotted vertical lines pinpoint the APV
Global ancestry characterization of Chilean children
Because genetic ancestry can affect pubertal growth phenotypes, it is important to account for individual European and/or Native American ancestries in the GWAS for PV, APV and HAPV (next section). Indeed, not accounting for Mapuche and Aymara sub-ancestries has shown to markedly underestimate associations with disease phenotypes, as compared to when these sub-ancestries are combined together (Lorenzo Bermejo et al. 2017).
To our knowledge, neither comparisons in PV, APV and HAPV between European and Native American children, nor comparisons in pubertal growth between Mapuche and Aymara children have been formally tested. Therefore, we decided to account for the effect of European ancestry as well as Native-American sub-ancestries in the GWAS linear regressions for PV, APV and HAPV. Hence, we performed a global ancestry inference with ADMIXTURE (Alexander and Lange 2011), using \(K=4\) ancestral populations. We chose this K value because it was able to clearly distinguish the Mapuche and Aymara subcomponents. As reference Native American populations, we used Mapuche and Aymara; as European and African reference populations we used CEU and YRI from 1000G, respectively (see Methods for details). Figure 2 shows the global ancestry proportions of each subject in our study, where each vertical line represents a child, and the colors the different ancestries. Individuals of the reference populations are also included. We found that the mean Native American ancestry proportion of our children is predominantly Mapuche (0.438), while their mean Aymara ancestry proportion is small (0.026). Also, our sample has 0.521 European and 0.015 African mean ancestry proportions, which is consistent with previous studies (Eyheramendy et al. 2015; Vicuña et al. 2020). Of note, admixture is homogeneous among individuals, with very few subjects having >80–90% of a single ancestry.

Global ancestry proportions. Each vertical line represents a subject and the colors the different ancestries. CHI: Chilean. The reference populations are MPU: Mapuche, AYM: Aymara, EUR: European; AFR: African

Genomic context of the genetic signal associated with PV in boys. Shown is the copy number variant esv3624175 harboring the significantly associated variants rs75297609, rs57205007 and rs7919344 as well as the pseudogene RN7SKP143. The image includes chromosome bands intercalated between black and white blocks with 10 Kb of size
GWAS of PV, APV and HAPV
We performed GWAS for PV, APV and HAPV. In the regression model, we adjusted for gender, Native American local ancestry, Mapuche global ancestry, genotype, an interaction effect between gender and genotype and an interaction effect between gender and Native American local ancestry. To increase the power to detect significant associations, all regressions were performed in boys and girls pooled together. Importantly, however, the interaction effects in our analyses allowed to separate the effects of the genotype and local ancestry on just boys, just girls or both (see Methods). In the analysis in boys, we found 2 variant alleles, rs75297609-T and rs57205007-G, achieving the genome-wide association significance threshold of P < 5 x 10\(^{-8}\) for PV (Table 1). These two variants are located close to the RN7SKP143 gene (Table 1), a pseudogene of the 7SK gene with unknown function. The 2 associated variants, as well as RN7SKP143, are in turn located within the copy number variant (CNV) esv3624175 (Fig. 3), and thus likely represent the same association signal. Indeed, the genotypes of rs75297609 and rs57205007 among individuals show a Pearson’s correlation coefficient of 0.78. rs75297609-T and rs57205007-G have 8 and 13 carriers, respectively, as well as allele frequencies of 0.004 and 0.007, respectively. Table 1 also shows 6 variants achieving nominal significance (P <10\(^{-6}\)).
We did not find significant associations for APV. However, one variant, rs148840332, located in the RP11-184E9.1 lincRNA gene, was associated with APV at P \(=\)3 x 10\(^{-7}\) (Supplementary Table 1). In the GWAS for HAPV we did not find associations achieving nominal significance (P <10\(^{-6}\); data not shown). In the analysis in girls (see Methods), we did not find significant associations for any phenotype. The Manhattan plots for the PV and APV GWAS are shown in Fig. 4 and Supplementary Fig. 1, respectively.

Manhattan plot for PV in boys. Genome-wide association P values, expressed as − log\(_{10}\)(P value). Highlighted in blue are the 2 variants achieving genome-wide significant associations (with the associated gene names within parentheses) as well as the genes most closely located to variants with P-value <10\(^{-6}\) (in black). Variants with − log\(_{10}\)(P-value) between 0 and 2 are not shown

Manhattan plot for PV in imputed SNPs from the region surrounding the copy number variant esv3624175. The region shown encompasses \(\pm 500\) Kb from rs75297609 and rs57205007. Its physical coordinates are 10:91521574-92510199. Genome-wide association P values are expressed as − log\(_{10}\)(P-value). Highlighted in blue is rs7919344-T, the significantly associated variant allele located within esv3624175. All variants mapping esv3624175 are labeled in blue. Variants with association P value \(<10^{-5}\) (Table 2) also map esv3624175 (not shown)

Effect of global Mapuche ancestry on APV and HAPV. The top and lower panels show how APV and HAPV vary as a function of global Mapuche ancestry proportions in boys and girls considered together. The regression line is labeled in red
To interrogate more variants for putative associations with PV, we performed genotype imputations of SNPs located within \(\pm 500\) Kb of the association signal harboring rs75297609 and rs57205007. We used the Trans-Omics for Precision Medicine (TOPMed) server (Taliun et al. 2021), which has a panel of 97, 256 samples from diverse ethnic backgrounds, including South Americans. We performed linear regressions adjusting for gender, Mapuche global ancestry, genotype and an interaction effect between gender and genotype. We did not include Native American local ancestry as covariate, since we lacked this information for imputed SNPs. In the analysis in boys, we found that the intergenic variant allele rs7919344-T was significantly associated with PV (P \(= 1 \times 10^{-9}\); Fig. 5 and Table 2). We also found 7 intergenic SNPs achieving nominal genome-wide significance (P \(< 10^{-6}\); Table 2), including the previously associated rs75297609-T variant allele. All of these variants seem to be part of the same peak (Fig. 5). Indeed, rs7919344 is located 399 bp from rs75297609 and 11.8 Kb from rs57205007. rs7919344-T has 7 carriers and an allele frequency of 0.004. In the analysis in girls, we did not find significant associations between PV and the imputed SNPs from chromosome 10.
Effect of genetic ancestry on pubertal growth phenotypes
We analyzed whether PV, APV and/or HAPV are influenced by global genetic ancestry proportions. We found that Mapuche Native American global ancestry had average effect sizes of – 0.73 years (P = 0.02) for APV and − 4.3 cm (P = 0.04) for HAPV in boys and girls combined (Fig. 6). Global ancestry did not have a significant effect on PV.
We also evaluated the effect of per-SNP wise local ancestry over PV and APV in non-imputed genotypes (HAPV was not analyzed due to an absence of strong GWAS associations). In the regression model, we measured the effect of Native American local ancestry and also the interaction term between genotype and Native American local ancestry (see Methods). The variant allele with the strongest genome-wide association with PV for the interaction term between genotype and Native American local ancestry was rs6699943-C (P \(= 3 \times 10^{-7}\); Table 1). We also found strong associations for the two significant variants in the GWAS for PV (P = 0.002 for rs57205007-G and P = 0.005 for rs75297609-T; Table 1).
Discussion
In this study, we aimed to better understand the genetic architecture of pubertal growth. We conducted GWAS on PV, APV and HAPV. The two former traits are critical antropometric markers of pubertal growth (Granados et al. 2015; Sovio et al. 2009). However, besides a few association studies between candidate genetic variants and PV in Europeans (Sovio et al. 2009), we are not aware of previous GWAS on PV, APV or HAPV.
Our GWAS on called and imputed SNPs detected a single genetic signal overlapping the copy number variant (CNV) esv3624175, which harbors the significantly associated variants rs75297609-T, rs57205007-G and rs7919344-T. This suggests that a causal variant linked to this region is driving the association with PV. This hypothesis is also supported by the high correlation across the genotypes of rs75297609 and rs57205007. The associated variants are located close to the RN7SKP143 pseudogene, which has unknown function. Even though the biological functions of the >200 7SK pseudogenes are unknown, some of them might have growth-related functions. For instance, on the same chromosome 10, RN7SKP167 has been GWAS-associated with growth-related phenotypes, including bone mineral density and hip bone size (P = \(4 \times 10^{-11}\)) (Zhang et al. 2020). However, most pseudogenes are not functional, so it is also possible that the three significant GWAS associations could result from these variants being in high linkage disequilibrium with causal variants located in or around other growth-related genes. Unfortunately, there is no recombination map for the Chilean population to test this hypothesis.
We found an intron variant allele of the LCOR gene, namely, rs113497890-G—also on chromosome 10—reaching a P value very close to genome-wide significance (P \(= 6.3 \times 10^{-8}\)). This gene has been GWAS-associated with adult height (P \(= 4 \times 10^{-20}\)) and heel bone mineral density (P \(= 1 \times 10^{-8}\)) (Kichaev et al. 2019), two phenotypes likely related with pubertal height growth. Increasing our sample size may augment statistical power to detect a significant association of the LCOR variant. It remains to be elucidated whether the main effects of the variants associated with PV on body growth would result from their neuro-endocrine actions through the hypothalamic–pituitary–gonadal (HPG) axis that triggers puberty (Bordini and Rosenfield 2011), or rather over local growth factors responsible for tissue growth. It is also unclear what biological mechanism underlies the sex-specific effects of the associated variants.
We analyzed the relationship between longitudinal pubertal traits and genetic ancestry. There are very few studies addressing this topic. An epidemiological study performed in a multiethnic US cohort showed that the mean APV is 13.7 years in boys and 12.1 years in females (Granados et al. 2015). Our results show that Chilean children have a lower mean APV (12.7 years in boys and 10.8 years in girls). However, since that study on US children did not consider genetic variables, it is not possible to estimate whether or not ancestry contributes to these differences. In our study of Chilean children, whose range for Mapuche ancestry varied from 0.25 to 0.94, we obtained that a difference in ancestry proportion led on average to a 0.73 years decrease in APV between a European and a Mapuche Native American boy. To our knowledge, we are the first to quantify how Native American genetic ancestry affects pubertal growth phenotypes, specifically PV, APV and HAPV. It is also important to recognize that differences in height growth and puberty milestones by ethnicity are not only determined by genetic ancestry, but also are influenced by social disparities among ethnic groups (Idossa et al. 2018).
Mapuche and Aymara diverged from their common ancestral population \(\sim\)8750 years ago (Lindo et al. 2018). Thus, it is possible that long-time exposure to different environments as well as demographic forces led to changes in their genetic diversity, which in turn could have differentially affected pubertal growth-related phenotypes in a population-specific manner. However, to our knowledge, comparisons in pubertal growth between Mapuche and Aymara children have never been formally tested. By accounting for Mapuche and Aymara global ancestries in the GWAS linear regressions instead of considering them as a single ancestral component, we obtained more reliable GWAS associations, since most of the Native American ancestry of the Chilean children is Mapuche. Noteworthy, we found that the strongest genome-wide association for the interaction between genotype and local ancestry is rs6699943-C, an intron variant allele of the GLUL gene, which has not been GWAS associated with height growth-related traits.
Unfortunately, we did not replicate our findings in an independent cohort, since there is no other longitudinal paediatric growth cohort with admixed Native American and European ancestries. European cohorts such as those that comprise the Early Growth Genetics Consortium (Middeldorp et al. 2019), are not useful for replication, since the three associated variant alleles are only found in Africans and in Latino populations (when considering populations from 1000G) (The 1000 Genomes Project Consortium 2015). An exception is IBS (Spaniards) from 1000G, where rs75297609-T and rs57205007-G are found with allele frequencies of 0.005 and 0.009, respectively (information retrieved from the Variant Effect Predictor (McLaren et al. 2016). None of these variants are present in our Mapuche or Aymara reference panels. Future research will be needed to determine the ancestral origin of the genetic signal harboring the three associated variants.
The results of our study highlight the importance of including genetic ancestry when performing GWAS in populations of admixed ancestries, such as Latinos or African Americans. Finally, since pubertal growth traits significantly vary with genetic ancestry, GWAS on understudied longitudinal growth traits hold great potential to identifying loci playing functional roles in particular continental populations.
Subjects and methods
Sample
We analized individuals of the “Growth and Obesity Chilean Cohort Study” (GOCS) (Corvalán et al. 2013). This is a longitudinal follow-up of 1195 individuals recruited in 2006 (aged 3.5 years old) at different childcare centers from the South East Area of Santiago, Chile. The population represents a middle-low socioeconomic level and all participants were singleton births occurring during \(2002-2003\), who had birth weights between 2500 and 4500 g, with no medical or mental conditions. Annual measurements were carried out at Instituto de Nutrición y Tecnología en Alimentos (INTA), Santiago, Chile, by trained dietitians, and since 2010 visits took place every 6 months. We used standardized protocols and height was measured with a portable stadiometer (Harpenden 603; Holtain LTD, Crosswell, UK) to the nearest 0.1 cm.
Genotyping
We used genome-wide data of 848 individuals from Tobar et al. (2019) and genotyped 105 additional individuals for the present study. Genotype data was obtained using the Infinium \(^{{\textregistered } }\) Multhi-Ethnic Global BeadChip (Illumina). Genotyping was performed in the Human Genotyping laboratory at the Spanish National Cancer Research Centre, a member of CeGen (PRB2-ISCIII). Raw intensity genotype files were loaded into GenomeStudio v2.0.3 (Illumina) and automatic clustering was performed. We removed 18 samples with call rate \(<0.98\) from the total of samples genotyped. We also excluded variants with heterozygous genotypes on the X chromosome for males and variants calling genotypes on Y chromosome for females. Using PLINK 1.9 (Purcell et al. 2007) we removed 10 additional samples based on gender mismatch, heterozygosity rate (variants with ±3 SD from the mean heterozigosity) and ancestry outliers. For relatedness, we randomly discarded 1 person from each pair with IBD/IBS>0.2, corresponding to a kinship relationship between second and third degree. Excluded variants had missing genotype data \(>5\)%, duplicated physical positions (one variant was kept from each duplicate pair) and deviations from Hardy–Weinberg equilibrium (HWE) (P \(< 1 \times 10^{-6}\)). A-T and C-G tranversions were also excluded to avoid inconsistencies with the reference human genome (build GRCh37). We also excluded 25 boys whose last measurements were taken before they were 12 years old. After applying these filters, we obtained a clean data set of 904 individuals and 774, 433 autosomal variants.
Local ancestry estimation
We used RFMix (Maples et al. 2013) to infer the local ancestry of genomic fragments in the 904 Chilean individuals. As reference populations for the local ancestry analysis we used the following populations from the 1000 Genomes Project (The 1000 Genomes Project Consortium 2015): Yoruba (YRI, \(n = 108\)) for African ancestry, Utah Residents with Northern and Western European Ancestry (CEU, \(n = 99\)) for European ancestry, and Peruvian (PEL, \(n = 85\)) for Native American ancestry. We merged all sets obtaining 1196 individuals and the 774, 433 SNPs. We inferred the gametic phase of individuals using Beagle 5 software (Browning et al. 2018), and the HapMap37 human genome build 37 recombination map. We used PopPhased -n 5 and –forward-backward parameters as recommended in the RFMix manual.
Global ancestry estimation
We estimated the global ancestry proportions with ADMIXTURE (Alexander and Lange 2011), using K = 4 ancestral populations. As reference Native American populations, we included 7 Mapuche individuals from Vidal et al. (2019) and 64 Aymara individuals from Lindo et al. (2018) and Crawford et al. (2017). As proxies for the European and African components, we used 99 individuals with Northern/Western European ancestry (CEU) and 102 Yoruba individuals from the 1000G Project (The 1000 Genomes Project Consortium 2015), respectively.
Genotype imputation
SNPs were imputed using the TOPMed Server (Taliun et al. 2021), which includes a panel of 97,256 deeply sequenced human genomes. Genotypes aligned to the GRCh37/h19 assembly were submitted into the server and imputations were run with default parameters. Imputed genotypes were converted from GRCh38 to GRCh37/h19 assembly using CrossMap (Zhao et al. 2014). Chain files as well as the reference genome were obtained from the UCSC Genome Browser (Kent et al. 2002). Imputed genotypes were filtered by a R-square value of 0.5. Indels were removed and assignment of Reference ID (RS number) was based on 1000G’s SNPs.
Longitudinal model
We estimated the growth curves for each individual in the sample through non-linear longitudinal mixed models; one model fitted to the girls and another model fitted to the boys, as implemented in the R package SITAR (Cao et al. 2018). The model takes the form \(y_{it}=\alpha _i+h(\frac{t-\beta _i}{exp(\gamma _i)})\), where \(\alpha _i, \beta _i\) and \(\gamma _i\) are random effects for individual i, h is a natural cubic spline function of age (t in the model) versus height, where \(y_{it}\) represents the height of individual i (\(i=1,\ldots ,n\)) at time t. The fitted curves for each individual were used to calculate peak height velocity (PV), age at peak height velocity (APV) and height at the age of peak velocity (HAPV) using the function getapv (Cao et al. 2018).
GWAS for PV, APV and HAPV
For each of the three phenotypes we performed genome-wide associations by implementing linear regression models. The model included boys and girls together. We adjusted for gender, Native American local ancestry at each SNP, global ancestry for each individual and the genotype for each SNP. Global ancestry refers to the proportion of global Mapuche Native American ancestry and local ancestry was measured for each child at each SNP, and it consists of an estimate of the number of alleles that originate from Native America. It takes values 0, 1 or 2. We also included in the model an interaction effect between genotype and local ancestry and an interaction effect between genotype and gender. The linear regression equation for the model was the following:
where \(X_1\) is a dichotomous variable that represents male gender with 0 and female gender with 1. \(X_2\) represents the proportion of global Mapuche Native American ancestry (this variable takes values between 0 and 1), \(X_{3j}\) represents the local Native American ancestry at SNP j (takes values 0, 1 or 2 that represent the number of alleles that originate from Native Americans), and \(X_{4j}\) corresponds to the additive genotype representation of SNP j. ph represents the phenotype that can take on the three phenotypes that we consider (PV, APV and HAPV) and the subscript i stands for measurements at individual i. Note that the genetic association of SNP j on female children is given by \(\beta _{4j}+\beta _{5j}\), while the association on male children is \(\beta _{4j}\). To identify significant genetic variants we performed three test of hypotheses for each phenotype and each SNP j: (i) \(H_0: \beta _{4j}+\beta _{5j}=0 \text{ vs } H_1: \beta _{4j}+\beta _{5j}\ne 0\); (ii) \(H_0: \beta _{4j}=0 \text{ vs } H_1: \beta _{4j}\ne 0\); (iii) \(H_0: \beta _{5j}=0 \text{ vs } H_1: \beta _{5j}\ne 0\).
To test associations between imputed SNPs and PV, we implemented a regression model similar to the aforementioned model, but we excluded the effect of Native American local ancestry. The linear regression equation for the model was the following:
The symbols of the covariates, their effects and the hypotheses tested were described in the previous model.
Variant and gene annotations
Variant annotations corresponding to the GRCh37 (hg19) assembly were retrieved with the web tool Variant Effect Predictor (VEP) from Ensembl (McLaren et al. 2016). They included the Sequence Ontology (SO) consequence type and Gencode biotypes. Upstream and downstream variants were defined as those located 10 Kb upstream or downstream of the gene, respectively. Intergenic variants were defined as those located >100 Kb upstream or downstream of the closest gene. We used HaploReg v4.1 (Ward and Kellis 2012) to identify the genes located closest to associated intergenic variants. Reported GWAS associations were retrieved from the NHGRI GWAS Catalog (Welter et al. 2014). Only variants achieving the genome-wide associations threshold of P \(< 5 \times 10^{-8}\) were considered. When more than one variant in a gene has been associated with the same phenotype, we reported the strongest association.
Availability of data and material
We can provide summary data upon request.
Change history
03 August 2021
A Correction to this paper has been published: https://doi.org/10.1007/s00439-021-02315-x
References
The 1000 Genomes Project Consortium (2015) A global reference for human genetic variation. Nature 526:68–74
Alexander DH, Lange K (2011) Enhancements to the admixture algorithm for individual ancestry estimation. BMC Bioinf 12:246
Biro FM et al (2006) Pubertal correlates in black and white girls. J Pediatr 148:234–40
Bordini B, Rosenfield RL (2011) Normal pubertal development: Part i: the endocrine basis of puberty. Pediatr Rev 32:223–9
Browning BL et al (2018) A one-penny imputed genome from next-generation reference panels. Am J Hum Genet 103:338–348
Cao Z et al (2018) New approaches to obtaining individual peak height velocity and age at peak height velocity from the SITAR model. Comput Methods Programs Biomed 163:79–85
Chan Y et al (2015) Genome-wide analysis of body proportion classifies height-associated variants by mechanism of action and implicates genes important for skeletal development. Am J Hum Genet 96:695–708
Corvalán C et al (2013) Obesity is positively associated with dehydroepiandrosterone sulfate concentrations at 7 y in chilean children of normal birth weight. Am J Clin Nutr 97:318–25
Cousminer DL et al (2013) Genome-wide association and longitudinal analyses reveal genetic loci linking pubertal height growth, pubertal timing and childhood adiposity. Hum Mol Genet 22:2735–47
Cousminer DL et al (2016) The genetics of pubertal timing in the general population: recent advances and evidence for sex-specificity. Curr Opin Endocrinol Diabetes Obes 23:57–65
Crawford JE et al (2017) Natural selection on genes related to cardiovascular health in high-altitude adapted andeans. Am J Hum Genet 101:752–767
Day FR et al (2015a) Shared genetic aetiology of puberty timing between sexes and with health-related outcomes. Nat Commun 6:8842
Day FR et al (2015b) Puberty timing associated with diabetes, cardiovascular disease and also diverse health outcomes in men and women: the uk biobank study. Sci Rep 5:11208
Elks CE et al (2010) Thirty new loci for age at menarche identified by a meta-analysis of genome-wide association studies. Nat Genet 42:1077–85
Euling SY et al (2008) Examination of us puberty-timing data from 1940 to 1994 for secular trends: panel findings. Pediatrics 121(Suppl 3):S172–91
Eyheramendy S et al (2015) Genetic structure characterization of chileans reflects historical immigration patterns. Nat Commun 6:6472
Granados A et al (2015) Relationship between timing of peak height velocity and pubertal staging in boys and girls. J Clin Res Pediatr Endocrinol 7:235–7
Herbison AE (2007) Genetics of puberty. Horm Res 68(Suppl 5):75–9
Idossa D et al (2018) Commentary: Race and ethnicity in biomedical research - classifications, challenges, and future directions. Ethn Dis 28:561–564
Kent WJ et al (2002) The human genome browser at ucsc. Genome Res 12:996–1006
Kichaev G et al (2019) Leveraging polygenic functional enrichment to improve gwas power. Am J Hum Genet 104:65–75
Lindo J et al (2018) The genetic prehistory of the andean highlands 7000 years bp though european contact. Sci Adv 4:eaau4921
Lorenzo Bermejo J et al (2017) Subtypes of native american ancestry and leading causes of death: Mapuche ancestry-specific associations with gallbladder cancer risk in chile. PLoS Genet 13
Maples BK et al (2013) Rfmix: a discriminative modeling approach for rapid and robust local-ancestry inference. Am J Hum Genet 93:278–88
McLaren W et al (2016) The ensembl variant effect predictor. Genome Biol 17:122
Middeldorp CM et al (2019) The early growth genetics (egg) and early genetics and lifecourse epidemiology (eagle) consortia: design, results and future prospects. Eur J Epidemiol 34:279–300
Mills MC, Rahal C (2020) The gwas diversity monitor tracks diversity by disease in real time. Nat Genet 52:242–243. https://doi.org/10.1038/s41588-020-0580-y
Parent A-S et al (2003) The timing of normal puberty and the age limits of sexual precocity: variations around the world, secular trends, and changes after migration. Endocr Rev 24:668–93
Paternoster L et al (2011) Adult height variants affect birth length and growth rate in children. Hum Mol Genet 20:4069–75
Purcell S et al (2007) Plink: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–75
Soliman A et al (2014) Advances in pubertal growth and factors influencing it: Can we increase pubertal growth? Indian J Endocrinol Metab 18:S53–62
Sovio U et al (2009) Genetic determinants of height growth assessed longitudinally from infancy to adulthood in the northern finland birth cohort 1966. PLoS Genet 5(3):e1000409
Styrkarsdottir U et al (2019) Gwas of bone size yields twelve loci that also affect height, bmd, osteoarthritis or fractures. Nat Commun 10:2054
Taliun D et al (2021) Sequencing of 53,831 diverse genomes from the nhlbi topmed program. Nature 590:290–299
Tobar HE et al (2019) Identification and functional analysis of missense mutations in the lecithin cholesterol acyltransferase gene in a Chilean patient with hypoalphalipoproteinemia. Lipids Health Dis 18:132
Vicuña L et al (2020) Post-admixture selection on chileans targets haplotype involved in pigmentation and immune defense against pathogens. Genome Biol Evol 12 (8):1459–1470
Vidal EA et al (2019) Whole genome sequence, variant discovery and annotation in mapuche-huilliche native south americans. Sci Rep 9:2132
Ward LD, Kellis M (2012) Haploreg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucl Acids Res 40:D930–4
Welter D et al (2014) The nhgri gwas catalog, a curated resource of snp-trait associations. Nucl Acids Res 42:D1001–6
Zhang H et al (2020) Pleiotropic loci underlying bone mineral density and bone size identified by a bivariate genome-wide association analysis. Osteoporos Int 31:1691–1701
Zhao H et al (2014) Crossmap: a versatile tool for coordinate conversion between genome assemblies. Bioinformatics 30:1006–7
Funding
This work was supported by ANID FONDECYT [3170038 and 11200324 to L.V.; 1190346 to V.M.; 1160833 and 1200146 to S.E., T.N. and JP.M; 1150416 and 1150486 to JL.S. ]. S.E., T.N. and L.V. were additionally supported by the Instituto Milenio de Investigación Sobre los Fundamentos de los Datos (IMFD). The genotyping, led by JL.S., was supported by grant PT13/0001/0005 of PE I + D + i 2013-2016, funded by ISCIII and ERDF (Fondo Europeo de Desarrollo Regional).
Author information
Authors and Affiliations
Contributions
S.E. conceived the project. SE, TN, LV, JPM, LO and FM analyzed the data. AP and VM collected data. JLS and SE raised funds for genotyping, JLS supervised the collection of DNA samples for genotyping. LV wrote the manuscript, with contributions from SE. All authors critically reviewed and accepted the final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing financial interests.
Consent to participate
Consent to participate and publish was obtained from parents or guardians and children assented to participate when they turned 7 years of age. The study was approved by the Scientific Ethics Committees of Instituto de Nutrición y Tecnología en Alimentos (INTA) and Pontificia Universidad Católica de Chile.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vicuña, L., Norambuena, T., Miranda, J.P. et al. Novel loci and Mapuche genetic ancestry are associated with pubertal growth traits in Chilean boys. Hum Genet 140, 1651–1661 (2021). https://doi.org/10.1007/s00439-021-02290-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-021-02290-3