
Abstract
CD4+ and CD8+ T lymphocytes mediate most of the adaptive immune response against tumors. Naïve T lymphocytes specific for tumor antigens are primed in lymph nodes by dendritic cells. Upon activation, antigen-specific T cells proliferate and differentiate into effector cells that migrate out of peripheral blood into tumor sites in an attempt to eliminate cancer cells. After accomplishing their function, most effector T cells die in the tissue, while a small fraction of antigen-specific T cells persist as long-lived memory cells, circulating between peripheral blood and lymphoid tissues, to generate enhanced immune responses when re-encountering the same antigen. A subset of memory T cells, called resident memory T (TRM) cells, stably resides in non-lymphoid peripheral tissues and may provide rapid immunity independently of T cells recruited from blood. Being adapted to the tissue microenvironment, TRM cells are potentially endowed with the best features to protect against the reemergence of cancer cells. However, when tumors give clinical manifestation, it means that tumor cells have evaded immune surveillance, including that of TRM cells. Here, we review the current knowledge as to how TRM cells are generated during an immune response and then maintained in non-lymphoid tissues. We then focus on what is known about the role of CD4+ and CD8+ TRM cells in antitumor immunity and their possible contribution to the efficacy of immunotherapy. Finally, we highlight some open questions in the field and discuss how new technologies may help in addressing them.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The biology of human T lymphocytes, for practical reasons, has been mostly defined with T lymphocytes purified from peripheral blood, and the study of circulating lymphocytes is still the basis of our conceptualization of T cell-mediated immunity. However, peripheral blood T lymphocytes represent only about 1–2% of the total T cell pool in the body [1]. Therefore, the great majority of T lymphocytes that are in lymphoid tissues or that infiltrate or stably reside in non-lymphoid tissues, which play pivotal roles in immune surveillance and organ homeostasis, have not been investigated thoroughly.
The existence of T lymphocytes stably residing in non-lymphoid tissues, named tissue-resident or resident memory T (TRM) cells, has been postulated about 20 years ago [2] and demonstrated 10 years later in mice by transplantation and parabiosis experiments [3,4,5]. In humans, the long-term persistence of T cells in tissues has been recently demonstrated in transplanted organs, such as lungs, intestine, and liver, in which donor T cells have been shown to reside for more than 1 year inside the transplant but have not been detected in peripheral blood of the recipients [6,7,8,9,10].
TRM cells can be either CD4+ or CD8+ lymphocytes, dwell in non-lymphoid tissues, patrol their surroundings, and can promptly respond when re-exposed to their antigens, providing protective immunity independently of T cells newly recruited from peripheral blood [11, 12]. Upon antigen re-exposure, TRM cells can trigger rapid adaptive immune responses and promote innate responses such as maturation of dendritic cells (DC) and activation of natural killer cells [13]. They can also provide bystander protection against antigenic unrelated pathogens and amplify the activation of a small number of cells to generate an organ-wide response [14].
These features enable TRM cells to potentially act as critical players in the immune surveillance against solid tumors, especially at the early stages of cell transformation. In fact, the clinical manifestation of tumors implies that cancer cells have evaded immune surveillance, including that from TRM cells. At this point, immune responses are mainly mediated by newly recruited effector T cells specific for tumor antigens [15] or viral antigens from infected tumor cells [16].
Compared with their protective function in infectious diseases, the precise role of TRM cells in antitumor immunity is not well characterized. Nonetheless, recent studies in mouse models have provided evidence for a relevant contribution of TRM cells in the immune response to different tumor types [17,18,19,20]. Moreover, several studies in human tumors have shown a positive correlation between the abundance of TRM cells and a better prognosis in cancer patients (summarized in [21]).
Here, we review the current knowledge on the anatomic compartmentalization of T lymphocytes, describing how TRM cells are generated during an immune response and what are the signals that guide first their migration from lymph nodes to non-lymphoid tissues and then their retention and maintenance in tissues. We then focus on what is known about the role of CD4+ and CD8+ TRM cells in antitumor immune responses and their possible contribution to the efficacy of immunotherapy. Finally, we highlight some open questions in the field and discuss how new technologies, especially single-cell techniques, may help in addressing them. It is important to keep in mind that studies on TRM cell function have mostly relied on investigation in animal models in vivo due to the difficulty of accessing and performing experiments on human tissue samples in vitro. That is why in this review we emphasize data on TRM cell function in humans.
Anatomic compartmentalization of T lymphocytes
Lymphocytes patrol tissues to surveil the body against the attack of pathogens. They do so by continuously trafficking between tissues and lymph nodes. T lymphocytes can enter non-lymphoid tissues exclusively through extravasation from arteries and exit through either efferent lymphatics or veins. T cells can reach lymph nodes from efferent lymphatics or from arterial blood going into high endothelial venules; they can then return to blood circulation through efferent lymphatics merging with the lymph ducts that empty lymph into venous vessels (Fig. 1).

T cell trafficking in lymphoid and non-lymphoid tissues. T cells enter non-lymphoid tissues exclusively through arterial blood and exit either with lymph to reach lymph nodes via the afferent lymphatic vessels, or with venous blood. T cells can enter lymph nodes also directly from the arterial circulation through the high endothelial venules (HEV) and return to the blood circulation through the efferent lymphatic vessels and the lymph ducts
Different T cell populations use combinations of chemokine receptors, integrins, and selectins to migrate and home into different lymphoid and non-lymphoid tissues. For instance, the entry into secondary lymphoid organs (SLOs) depends on cell surface expression of L selectin (CD62L) and CCR7 [22, 23], while display of the selectin P ligand (CLA) guides lymphocyte migration to the skin [24], and α4β7 integrin and CCR9 lead homing to the small intestine [25, 26]. Based on the expression of CCR7, a seminal study [27] defined in human blood two populations of memory CD4+ and CD8+ T lymphocytes with distinct homing potential and effector function. On the one hand, CCR7– T cells (named effector memory, TEM) upon activation display effector function; express receptors, such as CXCR3, CCR5, and CCR1, driving migration into inflamed tissues; and have shortened telomere length [27] suggestive of reduced self-renewal capacity of these cells. On the other hand, CCR7+ T cells (named central memory, TCM) upon activation did not display effector function, could efficiently stimulate dendritic cells, and express receptors (e.g., CD62L and CCR7) driving recirculation within blood and lymphoid tissues, but upon antigenic re-challenge, they can then differentiate toward TEM cells, thus displaying effector function and expressing receptors that drive migration into inflamed tissues. Subsequent studies in mice corroborated this concept by identifying TEM cells infiltrating non-lymphoid tissues [2, 28].
The expression of cell surface molecules that drive T lymphocytes toward specific tissues increases the efficiency of the immune surveillance by focusing migration of CCR7– TEM into sites of infection/inflammation rather than into healthy tissues [29].
Tissue-infiltrating lymphocytes differ from the circulating ones for the expression of surface markers but also for the acquisition of tissue-specific effector functions. Already 30 years ago, it was demonstrated in patients with chronic hepatitis C that intrahepatic and circulating T cell clones from the same person, specific for the same HCV protein, differed in terms of T cell receptor (TCR) sequences and in their ability to help IgA antibody production by B lymphocytes, indicating functional compartmentalization of T lymphocytes to the site of disease [30]. More recently, it has been shown by high-throughput screenings that, indeed, T lymphocytes infiltrating different human tissues are characterized by the combined expression of trafficking and functional markers that are specific to the tissue microenvironment [31, 32]. This feature adds another layer of complexity to the heterogeneity of T cell subsets from peripheral blood. The picture is further complicated by the observation that the composition of tissue-infiltrating lymphocytes varies over time during life [32, 33], possibly reflecting the history of antigenic exposure in different anatomic compartments.
These data imply that each tissue may have a “compartment code” and that T lymphocytes need to display the right password to migrate and home into that anatomic site, exert their effector function, and eventually stably reside or recirculate as memory cells to provide local or systemic long-term protection.
Generation of TRM cells
T lymphocyte activation requires cell-to-cell contact for antigen recognition and the subsequent clonal expansion or the display of effector function. Thus, the different anatomic compartmentalization of different T cells that privilege either cell division or cytokines secretion maximizes the efficiency of target identification, elimination, and establishment of immunological memory. Indeed, naïve T cells follow a relatively restricted recirculation from blood to SLOs where they can encounter antigens drained from tissues, thus reducing the area of immune surveillance needed to mount a primary immune response. CD4+ and CD8+ naïve T lymphocytes are activated in lymph nodes (LNs) by recognizing peptide antigens presented on the major histocompatibility complex (MHC) of DCs. Antigens can be transported from the non-lymphoid tissues into the LNs by migratory DCs or arrive as soluble molecules transported with the lymph and then collected and presented to T cells by LN-resident DCs. After activation by stimulatory DCs, naïve T lymphocytes start to proliferate and differentiate into effector cells that migrate, in response to chemotactic cues, to tissues where they carry out their effector functions. Once the antigen (e.g., pathogen or cancer cell) has been cleared, most (> 90%) of effector T cells die by apoptosis, while a small fraction of antigen-specific T cells persists as TCM, TEM, or TRM cells to provide systemic and local long-term memory [34, 35] (Fig. 2). Despite the extensive death of effector cells, the remaining memory T lymphocytes specific for a given antigen are numerically more represented than the initial naïve counterpart, and furthermore, they can perform faster responses and a broader immune patrolling, since, with TEM and TRM, surveillance is extended to non-lymphoid tissues.

T cell activation and migration to non-lymphoid tissues. (a) Antigens are presented to naïve T cells either by migratory DCs, which collect antigens in the peripheral tissues, mature, and migrate into the lymph nodes, or by lymph node-resident DCs that take up antigens transported in solution with the lymph. (b) Activated T cells exit from lymph nodes reaching first venous and then arterial blood and home into inflamed tissue chasing a gradient of inflammatory chemokines thanks to the expression of the corresponding chemokine receptors. Once in the proximity of the inflamed tissue, T cells adhere to the vascular endothelium first through weak interactions with endothelial selectins, such as E- and P-selectin, and then arresting their rolling by binding with integrins to endothelial adhesion molecules. Stable adhesion is followed by extravasation guided by the chemokine gradient and the upregulation of CD69 expression. (c) Upon activation naïve T cells start to proliferate and differentiate into effector cells, which migrate to peripheral tissues where they perform their effector function. Once the antigen has been eliminated, the large majority of effector cells die by apoptosis, while a small fraction of antigen-specific T cells persists as TCM, TEM, or TRM cells, which are characterized by a tropism toward different anatomic sites, to provide systemic and local long-term protection
Differentiation of TCM, TEM, and TRM cells is not clearly defined, as different models have been proposed which take into account strength and duration of TCR stimulation, and levels of inflammatory signals [36, 37]. In particular, the role of TCR stimulation strength in generating TRM cells is controversial [38,39,40,41,42] and further complicated by the observation that TCR::pMHC affinity and the signaling strength downstream of the TCR represent independent parameters [43]. On the contrary, there is growing evidence that a TRM cell fate is imprinted, at least in part, before the entry of effector T cells into peripheral tissues and possibly already during the priming of naïve T cells [44, 45]. In agreement with this hypothesis, stimulation by different DC subpopulations has been shown to instruct different tropisms of activated T cells. Mouse studies demonstrated that naïve T cell activation primarily occurs in SLOs proximal to the tissue of antigen sampling and that the tissue-emigrating DCs convey, together with the antigen, signals to polarize the migration of the activated T lymphocytes to the same non-lymphoid tissue they came from [26, 46, 47]. For instance, DCs migrating from the intestine can produce retinoic acid and TGF-β to induce the expression of the gut-homing molecules α4β7 and CCR9 on T cells [48, 49]. Similarly, cutaneous and lung DCs have been shown to instruct T cell homing to skin and lungs by inducing the expression of CCR10 and CCR4 on the T cell membrane [46, 50]. On the contrary, soluble antigens drained in lymph nodes from the afferent lymph and presented by LN-resident DCs can induce the differentiation of TCM-like Tcf1+ T cells, which are endowed with higher self-renewal capacity [51] and imprinted to recirculate within SLOs. Recently, a specific population of human migratory CD1c+ CD163+ DCs (also named DC3) has been shown to induce in vitro the generation of CD8+ cells with a TRM signature through TGF-β signaling [52, 53]. The CD8+ TRM cells generated were able to colonize and inhibit the progression of breast cancer in a humanized mouse model [52]. Moreover, DC3 cells were found to infiltrate human luminal breast cancer primary tumors and their frequency correlated with that of CD8+CD69+CD103+ TRM cells [53], supporting their role as TRM cell inducers. The hypothesis that the tropism of memory T cells is dictated by the nature of the stimulating antigen-presenting cells (APCs) and by the site of antigen inoculation is corroborated by the observation that some vaccination strategies can preferentially induce the generation of TRM cells. For instance, the intranasal administration of a live-attenuated influenza vaccine, but not the intraperitoneal administration of an injectable inactivated influenza virus, induced the formation of virus-specific lung CD4+ and CD8+ TRM cells, in mice. These virus-specific TRM cells mediated protection independently from circulating T cells and could also provide heterosubtypic immunity [54].
Together these data indicate that naïve T lymphocytes can be imprinted to form a pool of TRM cells depending on signals received during activation in LNs, signals that include the nature of the stimulating DCs, exposure to TGF-β, and possibly the strength and the duration of activation.
A critical step in the generation of TRM cells is the recruitment of the activated T cells into the tissue. Effector T cells, differentiated from recently activated naïve T cells, enter tissues chasing chemotactic inflammatory signals released by the diseased site, such as CCL5 and CXCL9 or CXCL10 that attract activated T cells expressing CCR5 and CXCR3, respectively [55,56,57,58]. However, activated T lymphocytes can transiently acquire the ability to enter tissues in the absence of inflammation [4, 59] which can broaden the immune surveillance beyond the site of initial antigen encounter. It has been hypothesized that TRM and effector T cells differentiate independently from different precursors branching early upon naïve T cell activation, since CD8+ TRM cells have been shown to develop from tissue-infiltrating T lymphocytes that do not express the effector T cell marker KLRG1 [58, 60]. However, this hypothesis has been challenged by the finding that in vivo a sizeable fraction of KLRG1– memory T cell precursors derive from effector T cells that have downregulated KLRG1 expression and that these “exKLRG1” precursors can generate TRM cells [61]. The hypothesis TRM cells can differentiate from effector T cells is further supported by the identification of effector T cells bearing a TRM-like transcriptional signature [44] and by the finding that TRM cell differentiation relies on a hybrid transcriptional program regulated by both effector- and memory-specific transcription factors. While mouse TRM cell differentiation is regulated by Blimp-1, its homolog Hobit, Runx3, Id2, and Id3 [19, 62, 63], there is no evidence that these transcription factors dictate the transcriptional identity of human TRM cells. Although Runx3 has been reported to regulate (in cooperation with Notch signaling) the transcriptional program of both mouse and human TRM cells [19, 64,65,66], there is no evidence of a master transcriptional regulator of human TRM cells, and additional investigation is necessary to understand how the different steps of TRM cell generation and maintenance are controlled.
These data indicate that TRM cell precursors enter peripheral tissues as memory precursor effector cells derived from the activation of TRM-fate imprinted naïve T cells.
Tissue retention and maintenance of TRM cells
Once inside tissues, TRM cell precursors need to receive retention signals that make them long-lived resident cells. In mice, CD69 upregulation is one of the first events that allow T lymphocytes to extravasate, enter, and persist in tissues [67]. CD69 post-transcriptionally antagonizes the sphingosine-1-phosphate receptor (S1PR1) [68], which regulates chemotaxis toward the sphingosine-1-phosphate (S1P) produced by the endothelial cells of lymph and blood vessels [69]. CD69 interacts with S1PR1 transmembrane and membrane-proximal regions and the helix 4, resulting in S1PR1 internalization and degradation without immediate effects at the transcriptional level [68]. Later, S1PR1 is also transcriptionally silenced in parallel with the downmodulation of its transcriptional regulator KLF2 [70]. Cytokines that can induce CD69 upregulation and S1PR1 downregulation include type-I interferons [71], TNF-α, and IL-33 [70]. The same mechanism of tissue retention mediated by the CD69-S1PR1 antagonism takes place in the lymph nodes where, after priming by APCs, recently stimulated T lymphocytes upregulate CD69 to transiently prolong their stay and allow full activation. For this reason, CD69 upregulation is also an early activation marker [72]. In addition to S1PR1, other sphingosine receptors can guide leukocytes toward lymph and blood vessels [73]. Among these, S1PR5 has been recently shown in the mouse to contribute to the tissue egress of antigen-experienced CD8+ T cells, under the transcriptional control of T-bet and Zeb2 transcription factors [74].
While CD69 expression, which limits the S1PR1 signaling and prevents T cell egress from tissues, is common to activated T cells that enter peripheral tissues, the integrins CD103 (αE integrin) and CD49a (α1 integrin) are expressed in response to TGF-β and IL-12 signaling [75, 76] and function as anchors to retain T cells within the tissue, so making them resident T cells [60, 77,78,79]. By binding to E-cadherin and collagen IV, respectively, CD103 (in the form of αEβ7 integrin) and CD49a (in the form of α1β1 integrin) not only anchor TRM cells to the tissue but can also enhance their proliferation and limit apoptosis, thus providing survival signals [80,81,82]. In addition to CD103 and CD49a, also CXCR6 contributes to the localization and the maintenance of TRM cells by binding to CXCL16 present on epithelial cells membrane and triggers conformational changes in integrins that support cell adhesion [83,84,85]. Additional molecules contributing to tissue retention of TRM cells are RGS1 and RGS2 [64, 86] which accelerate the termination of heterotrimeric G protein signaling and, in turn, reduce the sensitivity to chemotactic cues [87], especially in the gut [88]. The imprinting during the priming phase may be sufficient to sensitize TRM cell precursors to upregulate tissue retention molecules in response to cytokines in the tissue microenvironment, such as TGF-β and IL-12, but local re-encounter of their antigen can enhance TRM cells proliferation and tissue retention [89, 90].
In summary, data indicate naïve T cells can be preconditioned in homeostasis and imprinted during activation by migratory DCs to become TRM cells through TGF-β signaling. Moreover, additional signals in tissues, such as re-encounter of their antigen and presence of cytokines such as type-I interferon, IL-33, TGF-β, and IL-12, will define whether the memory cell precursors will be retained in tissues and will differentiate into long-lived TRM cells (Fig. 3).

TRM cell differentiation. Naïve T cells can be reversibly preconditioned in homeostasis and imprinted during activation by migratory DCs to become TRM cells through TGF-β signaling. Migratory DCs can also instruct T cells to home into the same non-lymphoid tissue they came from. For instance, intestinal and cutaneous migrating DCs, by metabolizing respectively vitamin A and vitamin D, induce the expression of chemokine receptors guiding activated T cell homing into the gut and the skin. Recently activated effector T cells reach the inflamed non-lymphoid tissues through arterial circulation and extravasate also thanks to the competition between CD69 and S1PR1. Here, a subset of effector TRM precursor cells upregulates the expression of tissue-retention molecules (e.g., CD103, CD49a, and CXCR6) in response to TGF-β and IL-12, differentiating into long-lived TRM cells. Re-encountering the cognate antigen presented by professional and non-professional antigen-presenting cells may enhance the establishment of TRM cells
It is worth noting that signals required to become TRM cells may diverge in different tissues [91] and their systematic identification is still an open area, especially in humans.
Markers for the identification of TRM cells
The expression of CD69, CD103, and the other mentioned tissue retention molecules (Table 1) has been used in different combinations to identify TRM cells. However, none of the described proteins is sufficient alone to define TRM cells, nor is the contemporary expression of all of them necessary. For instance, CD69 expression alone, which has been often used in the literature for the identification of TRM cells, is not sufficient to univocally define T cells residing in tissues as its upregulation represents a common feature of T lymphocytes leaving the circulation and entering peripheral tissues, including those that are not going to persist there. Such complexity in the expression patterns of surface markers may reflect differences between CD4+ and CD8+ TRM cells and the presence of tissue-specific signals or the occupation of non-overlapping sub-anatomic niches [92,93,94]. For instance, CD4+ TRM cells in the human dermis lack CD103 expression, whereas those in the epidermis are CD103+ [92]. Also, CD8+ CD103+ and CD103– TRM cells are found in the human intestine, where they show different preferential distribution and effector potential: both the populations infiltrate the intestinal epithelium and lamina propria, but the CD103+ TRM cells are more frequent in the epithelium and express higher IL-2 and IL-7Rα, while CD103– TRM cells are more frequent in the lamina propria and produce higher granzyme K [95].
Here, we propose to define TRM cells based on the expression of at least one of the molecules mediating tissue retention, i.e., CD103, CD49a, and CXCR6, and on the simultaneous lack of molecules, such as S1PR1 and S1PR5, which guide T cell egress from tissues (Table 2). This combination of markers cannot inform on the long-term tissue retention of TRM cells but provides a snapshot of the cell status at a defined time point without excluding the potential of leaving non-lymphoid tissues under different circumstances. In any case, there is evidence that TRM cells re-entering the circulation upon reactivation are biased to return to the tissue of origin and become again resident T cells [96].
As for the identification of tissue-specific signals driving TRM cell differentiation, a comprehensive definition of cell markers that identify CD4+ and CD8+ TRM cells at different anatomic and sub-anatomic sites is still missing.
TRM cells in tumor immunity
Accumulating evidence shows that TRM cells frequently reside in human tumors, especially those of epithelial origin, and can have a protective function [21, 97, 98]. How TRM cells participate in controlling tumor growth has not been determined yet, nor is the contribution of tumor-specific or bystander TRM cells clear. The pool of intra-tumor TRM cells can be made of lymphocytes present in the tissue before the appearance of transformed cells, such as those elicited by microbes and not specific for tumor antigens, and of T cells specific for tumor-derived antigens. Tumor-specific TRM cells can be generated following the effector response against immunogenic transformed cells (Fig. 4). During the equilibrium phase of tumor development, they can kill transformed cells and contribute to the control of tumor growth [99] (Fig. 5). However, clinical manifestation of tumors implies that cancer cells have evaded immune surveillance, for instance by inducing T cell dysfunction. Dysfunctional TRM cells can be reactivated by blockade of inhibitory checkpoints (e.g., CTLA4 and PD1) and they can contribute to the efficacy of immunotherapy (see below). Nevertheless, de novo immune responses triggered by tumor neoantigens made available by radio- or chemotherapy-induced immunogenic cell death [100] are usually mediated by newly generated effector T cells (Fig. 5). In this context, new tumor-specific activated T cells can migrate from LNs in blood circulation and then into cancer tissue where they can exert effector function [15]. Consistently with this scenario, we observed in CRC patients treated with neoadjuvant chemotherapy that the T cells that infiltrate CRC liver metastasis and show the highest clonal expansion are effector T cells. These effector T cells show evidence of TCR-mediated activation and lack typical features of TRM cells (S.N. and S.A, unpublished), suggesting they have been recently recruited from circulation. Longitudinal studies would be required to assess whether such effector responses are able to generate memory T cells and if these cells could become resident due to tumor microenvironmental factors, such as TGF-β.

Generation of tumor-specific effector and memory T cells. Tumor antigens released by cancer cells are collected by dendritic cells and presented to naïve T cells in lymph nodes. Differentiated tumor-specific effector T cells migrate to the tumor site where they perform their effector function. Effector CD8+ and CD4+ T cells exert their anti-tumor activity by directly killing tumor cells and providing help to other immune cells, while TREG and TR1 cells suppress the immune response. Immune-mediated cancer cell death can induce the release of more tumor antigens and sustain anti-tumor immunity

Tumor and immune system co-evolution. Tumor-specific TRM cells can be generated following the effector response against the first immunogenic cancer cells. During the equilibrium phase of tumor development, TRM cells can kill cancer cells and contribute to the control of tumor growth. However, the tumor can escape immune surveillance, including that from TRM cells, and generate an immunosuppressive microenvironment. Chemotherapy and immunotherapy can reactivate the anti-tumor immune response: chemotherapy by inducing the release of tumor neoantigens, which can trigger the generation of new tumor-specific effector T cells; immunotherapy by reactivating the effector function of dysfunctional memory T cells
CD8+ TRM cells
The abundance of tumor-infiltrating CD8+ CD103+ T cells and the elevated expression of a TRM signature correlate significantly with prolonged disease-free or overall survival of patients with many different tumor types, including breast cancer [101, 102], lung cancer [17, 103,104,105], colorectal cancer (CRC) [106], melanoma [15, 107, 108], head and neck cancer [109], esophageal cancer [110], gastric cancer [111], hepatocellular carcinoma [112], ovarian cancer [113,114,115], cervical cancer [116], endometrial adenocarcinoma [117], and bladder cancer [118]. Also, the localization of TRM cells within the tumor matters. Indeed, the infiltration of CD103+ T cells in the tumor epithelium, but not in the tumor stroma associates with longer survival, as reported in breast [101, 102] and lung [104] cancers. The prognostic power associated with higher TRM cell abundance might vary depending on the tumor stage and is higher in early-stage disease when TRM cells are probably still functional.
The abundance of CD8+ CD103+ T cells has been negatively correlated with the presence of metastasis in patients with gastric adenocarcinoma [111], but the role of TRM cells in protecting from cancer spreading has to be clarified yet. Nonetheless, recent evidence from a mouse model of melanoma demonstrated that TRM cell activation can trigger the maturation and migration of dermal DCs to LNs, and induce new cytotoxic CD8+ T cell responses that suppress the growth of the primary tumor and of melanoma cells disseminated to the lung [15].
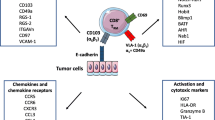
Tumor-specific CD8+ TRM cells can directly kill tumor cells in a MHC-I-restricted manner [109] through the production of cytotoxic molecules. Indeed, they can express high levels of granzyme B, perforin, and the degranulation marker LAMP-1 (CD107a) [103, 105, 119]. Notably, TRM cells in ovarian cancer show features of activated T cells, such as an elevated surface expression of HLA-DR coupled to the downregulation of CD127 (IL-7Rα), and the co-expression of the proliferation marker Ki67 [113]. Both CD103+ and CD49a+ CD8+ TRM lymphocytes are potent killer cells [107]. CD103 is recruited at the immunological synapse and provides co-stimulatory signals [120]. Its interaction with E-cadherin on target cells promotes the phosphorylation of ERK1/2 and phospholipase Cγ1 in TRM cells and is required to polarize the exocytosis of cytolytic granules and optimize the killing of the target [119, 120]. However, CD49a+ TRM cells have been reported to have a superior protective function than CD103+ ones, at least in a mouse model of airway viral infection, possibly due to their relatively higher local motility that allows a more efficient patrolling of the tissue [121].
At the same time, tumor-specific TRM cells can promote antitumor immunity through the release of cytokines and chemokines that help activation or recruitment of other immune cells [15]. Indeed, in mice, effector cytokines released by TRM cells can stimulate local DCs, natural killer cells, and other T cells [13, 122, 123].
TRM cell effector function also depends on the nature of the stimulating APC. In principle, CD8+ TRM cells can be activated by APCs of non-hematopoietic origin, including tumor cells, or by APCs of hematopoietic origin through antigen cross-presentation. In a mouse model of viral infection, lung CD8+ TRM cell activation by professional or non-professional APCs resulted in different functional outputs: antigen presentation by non-hematopoietic APC induced TRM cells proliferation and the expression of proinflammatory cytokines, likely to eliminate infected cells; on the contrary, antigen presentation by hematopoietic APCs elicited TRM cells to express several interferon-responsive genes but limited the production of some pro-inflammatory cytokines and chemokines, such as Ccl3, Ccl4, Ifng, and Xcl1, possibly to prevent excessive local inflammation and leukocytes recruitment [124].
Collectively, the described features indicate CD8+ TRM cells can display anti-tumor effector responses, but additional investigation is still needed to dissect the role of CD8+ TRM cell subsets expressing different tissue retention molecules, such as CD103, CD49a, and CXCR6.
CD4+ TRM cells
Although CD4+ TRM lymphocytes that accumulate in tissues often outnumber CD8+ TRM cells [125], most studies on TRM cells have focused on CD8+ T cells and the understanding of CD4+ TRM cell biology is less advanced. Tissue-resident CD4+ TH1, TH2, TH17, and regulatory T cells (TREG) have been identified in many tissues where they have been shown to protect against pathogens but also to exacerbate inflammatory and autoimmune diseases [126,127,128,129]. Instead, the functional dissection of CD4+ TRM cells has been complicated by their heterogeneity [31].
The knowledge gap of CD4+ TRM cells is even bigger in the context of anti-tumor immunity. In the last decade, growing evidence has indicated that CD4+ T cells can contribute to control tumor growth in humans [130,131,132,133,134], and CD4+ T cells specific for neo-antigens have been detected within tumors with elevated Tumor Mutational Burden, such as melanoma [132]. CD4+ T cells are necessary for the priming and differentiation of CD8+ T cells [135] and support durable tumor-specific cytotoxic T cell responses by instructing the downregulation of coinhibitory receptors and enhancing the capacity of CD8+ T cells to infiltrate tumors [136]. Moreover, CD4+ T cells can cooperate with cytotoxic T lymphocytes in bystander killing of cancer cells [137] and can also acquire cytotoxic function so as to kill tumor cells expressing MHC-II as effectively as CD8+ T cells [138, 139]. In addition, CD4+ T cells with a TFH-like phenotype (CXCR5+/PD-1+/CXCL13+) colocalize with B lymphocytes in tertiary lymphoid structures (TLSs) that are particularly present within non-epithelial tumors, such as melanoma and sarcoma, and the presence of these TLSs correlates with better responses to immunotherapy and longer patients survival [140, 141].
TH17 cells have been reported to infiltrate human ovarian and colon cancer [142, 143]. Tumor-infiltrating TH17 cells produced cytokines, such as CXCL9 and CXCL10, contributing to the recruitment of effector T cells. In the context of ovarian cancer, the abundance of TH17 cells correlated with patient survival and was reduced in more advanced stages [143]. Although in this study tumor-infiltrating TH17 cells were not tested for the expression of TRM cell markers, they were shown to express several integrins and to be long-lived. Moreover, their resistance to apoptosis, mediated by Bcl-2 expression, depended on Hif-1α and Notch [142], two transcription factors involved in the regulation of human TRM cell phenotype. Notably, IL-17-producing CD4+ TRM cells have been found in the lung of patients infected with Mycobacterium tuberculosis and SARS-CoV-2 [129, 144], and in transplanted intestines [95]. Moreover, fate-mapping experiments using an IL-17A-reporter mouse model, combined with the analysis of clonal distribution by TCR sequencing, demonstrated that a significant fraction of lung CD4+ TRM cells, including IL-17-negative ones (exTH17 TRM cells), derive from effector TH17 cells upon Klebsiella pneumoniae infection [145]. These data suggest that TH17 cells may represent one of the major CD4+ TRM cell populations at mucosal sites, possibly guided in their homing by the chemokine receptor CCR6. Supporting this hypothesis, a subset of TH17 cells expressing upon activation the immunoregulatory cytokine IL-10 and tissue residency-associated molecules, such as CD69, CXCR6, and ITGB7, has been identified in the circulation of human subjects, possibly representing TRM cell precursors [146].
More recently, CD4+ CD103+ TRM cells infiltrating human non-small cell lung cancer (NSCLC) were shown to produce IFN-γ and TNF-α, resembling a TH1 phenotype. The authors suggested that the IFN-γ produced by CD4+ TRM cells could contribute to attracting CD8+ TRM cell precursors and showed that the frequency of the two populations infiltrating NSCLC positively correlated [147].
TREG lymphocytes abundantly infiltrate CRC, NSCLC, and breast cancer and have a distinct profile from circulating TREG cells [148, 149]. The abundance of tumor-infiltrating TREG (TI-TREG) cells is associated with a poor prognosis in human cancer. Although TI-TREG cells do not show enhanced expression of TRM cell markers compared to other tumor-infiltrating T cell populations, they constitutively express high levels of CCR8 [148, 149], which can mediate tissue retention in epithelial tissues and has been reported as a feature of TRM cells in human skin [150].
Another population of suppressive CD4+ T cells, characterized by the lack of FOXP3 and increased GZMK expression, named TR1 cells, has been identified to infiltrate different human solid tumors. Based on single-cell TCR sequencing analysis, TR1 cells are clonally unrelated to FOXP3+ TREG cells and clonally expanded in tumors. Their abundance, measured by the expression of the specific marker CHI3L2, negatively correlates with patients’ survival but positively correlates with the response to immunotherapy [151]. Despite tumor-infiltrating TR1 cells were not specifically tested for the expression of genes characteristic of TRM cells, they express CCR5 [152], which is required for the migration toward inflamed tissues and can contribute, in combination with CCL5 locally produced by macrophages, to CD4+ TRM cells tissue retention [55, 153].
In conclusion, more investigative efforts are needed to dissect the phenotypic and functional heterogeneity of CD4+ TRM cells and to better understand their role in anti-tumor immunity.
TRM cells in immunotherapy
The development of single-cell sequencing technologies coupled with TCR sequencing has provided a new powerful tool to deeper investigate the heterogeneity of tumor-infiltrating lymphocytes. Indeed, TCR represents the identity card of T lymphocytes and knowledge of its sequence allows to track individual T cell clones in different tissues. Several studies have investigated at the single-cell resolution the transcriptional identity, and sometimes clonal distribution, of T cells infiltrating human tumors, including breast cancer [154, 155], NSCLC [156], CRC [157], hepatocellular carcinoma [158], bladder cancer [159], head and neck cancer [160], and basal cell carcinoma [161, 162]. However, only some of these studies have taken into consideration phenotypes specifically associated with tumor-infiltrating TRM cells. The most relevant feature of these TRM cells, identified as CD8+ CD103+ T cells, common to breast, lung, and basal cell cancers, was an elevated expression of genes coding for immune checkpoint receptors, such as PDCD1 (PD-1), CTLA4, HAVCR2 (TIM-3), TIGIT, and LAG3, and a higher expression of GZMB (granzyme B) and PRF1 (perforin) compared with CD8+ CD103– T cells [155, 156, 161, 163]. A subset of the CD8+ TRM cell population also expressed genes associated with cell proliferation. Moreover, in lung cancer, a CD8+ CD103+ T cell subset with low PD-1 expression has been also identified and proposed to represent a “pre-exhaustion” state [156]. The expression of immune checkpoint receptors by tumor-infiltrating human CD8+ TRM cells has been confirmed at the protein level by several independent studies [17, 103, 105, 113, 117, 164].
The presence of proliferation markers and the identification of TRM cell subsets with a graded expression of immune checkpoint receptors suggests that TRM cells may include tumor-specific T cells that have become dysfunctional upon chronic stimulation in the tumor microenvironment. Interestingly, immunotherapy with anti-PD-1 boosts the formation of TRM cells in an adoptive cell transfer protocol in melanoma-bearing mice [20]. In humans, the number of infiltrating CD8+ CD103+ TRM cells significantly increases in melanoma patients early upon PD-1 blockade, especially in patients responding to immunotherapy [108]. Similarly, in NSCLC patients, the density of CD8+ CD103+ TRM cells before immunotherapy positively associates with improved outcomes and increases during the therapy in most of the responders, but not in non-responder patients [165]. Also, in esophageal squamous cell carcinoma, CD8+ CD103+ TRM cells show enhanced proliferation and cytotoxic potential, measured by Ki67 and CD107a staining, compared to tumor-infiltrating CD8+ CD103– cells after anti-PD-1 therapy [110]. Notably, PD-1 blockade on CD8+ TRM cells, freshly isolated from human lung cancer, promoted a strong MHC-I-restricted cytolytic activity against autologous tumor cells ex vivo, which was impaired by blocking CD103 interaction with target cells [103]. These data indicate that immunotherapy unleashes the effector capacity of TRM cells and possibly induces their proliferation. Whether the increase in TRM cell infiltration upon PD-1 blockade is also sustained by the generation of new TRM cells remains to be addressed.
The expression of immune checkpoint receptors is present also in TRM cells from normal tissues [58], but the fact that tumor-infiltrating TRM cells are less capable to produce effector molecules suggests the acquisition of a deeper state of dysfunction [166], which may be one of the reasons why cancer develop despite the presence of tumor-specific TRM cells. Altogether, data from the literature indicate that CD8+ CD103+ TRM cells include tumor-specific exhausted T lymphocytes that can be revitalized by immunotherapy. Therefore, inducing a pool of tumor-specific TRM cells, for instance by specific vaccination strategies [17], combined with immunotherapy may represent a possible therapeutic strategy to promote tumor regression.
TRM cell maintenance in the tumor microenvironment
Considering that TGF-β can be highly expressed in the tumor microenvironment and that it regulates TRM cell formation and maintenance [167, 168], it is not surprising that tumors are frequently populated by TRM cells. Also, hypoxia, another environmental cue characterizing the tumor microenvironment has been recently shown to synergize with TGF-β to induce a TRM phenotype on in vitro stimulated human CD8+ T cells [169].
Differently from effector T cells that mainly rely on glucose and glutamine metabolism [170], TRM cells express high levels of the free-fatty acid (FA) transporters Fabp4 and Fabp5 and catabolize short-chain free-FA as a preferential energy source [171]. Relying on FA metabolism could give an advantage to TRM cells by limiting the competition for nutrients with tumor cells, which usually consume glucose and create local hypoglycemia [172]. Indeed, promoting FA metabolism with a PPAR-α agonist sustains CD8+ T cell effector function, delays tumor progression, and enhances the therapeutic efficacy of PD-1 blockade in a mouse melanoma model [173]. In turn, immune checkpoint blockade promotes Fabp4 and Fabp5 expression in TRM cells, but not in tumor cells, promoting lipid uptake and improving their survival [111]. Moreover, TRM cells from melanoma and gastric adenocarcinoma patients show an increased FA uptake compared to the CD103– infiltrating and the circulating T lymphocytes [111, 173]. However, since FA oxidation requires oxygen consumption, the lipid metabolism under hypoxic conditions usually leads to lipogenesis, making the lipid storage an inaccessible energetic source until reoxygenation. It is possible that, differently from acute hypoxia, in chronic limiting oxygen conditions, cells may reset their metabolism and prioritize the oxidation of FA and glutamine over pyruvate using the spare oxygen available [174]. Nonetheless, it remains to be addressed if and how T cells in the tumor microenvironment metabolized intracellular lipid storages for energy generation or if TRM cells can adapt their metabolism to exploit other nutrients available in different tumor niches.
Targeting metabolism to boost T cell anti-tumor activity is a field of active investigation. For instance, T lymphocytes with increased intracellular L-arginine levels display enhanced survival and anti-tumor effector capacity [175], and tumor colonization with an Escherichia coli strain engineered to continuously convert ammonia to L-arginine increases the number of tumor-infiltrating T cells and synergizes with PD-L1 blockade in the clearance of tumors, in mice [176]. Together these data indicate that manipulation of T cell (including TRM cell) metabolism represents a new area of immunotherapy.
Conclusions
TRM cells constantly patrol tissues and their adaptation to the local microenvironment makes them professional tissue defenders. Current knowledge from mouse and human studies supports a role for TRM cells in anti-tumor immunity, especially in the first stages of tumor development, and growing evidence suggests they may enhance the efficacy of immunotherapy, highlighting their possible contribution to controlling tumor growth also after its clinical manifestation.
So far, analyses of TRM cells in human tumors have mostly relied on the expression of one (e.g., CD103) or a few markers, but it is evident that TRM cells expressing the combination of different molecules may represent subsets occupying different anatomic niches and having different patrolling and effector capacity. Moreover, research efforts have largely focused on CD8+ TRM cells, while our understanding of CD4+ TRM cell function is still very limited. The anti-tumor activity of TRM cells may change depending on the tissue of origin and on tumor mutational load, and the role of TRM cells in protecting from the metastatic spread remains unclear. Thus, we need to systematically dissect the function of the different CD4+ and CD8+ TRM cell subsets from different tissues and from various primary and metastatic tumors.
The development of high-throughput technologies, including multiparametric flow and mass cytometry, microscopic tissue spatial analysis, and single-cell RNA and TCR sequencing, will certainly help in gaining novel insights into the biology of human TRM cells isolated from clinical material. For instance, combining the information from single-cell RNA and TCR sequencing allows for dissecting the phenotype and the clonal expansion of T cells specifically infiltrating tumor tissues and the identification of expanded T cell clones, which may include tumor-specific T cells. Knowing the proportion of intratumor-expanded TRM cells compared to other infiltrating T cell populations may reveal the relative contribution of circulating and resident T lymphocytes in anti-tumor immunity, while the dissection of their transcriptional profile may inform about the contribution of T cell subsets with different identities and the activation or silencing of specific signaling pathways. However, without prior knowledge of the tumor antigens, we are still not able to distinguish in this kind of analysis which are, among the expanded T cell clones, the tumor-specific T cells. Although the expression of CD39 has been proposed to identify tumor-specific T lymphocytes [109, 177], its expression may simply tag chronically stimulated T cells that become exhausted. These cells are likely to be enriched in tumor-specific T cell clones in the tumor microenvironment, but CD39– tumor-specific T cells have been also identified and shown to have a higher proliferation capacity upon re-challenge compared to the CD39+ T cells [178]. Thus, the identification of better markers associated with tumor-specific T cells would be extremely valuable.
In addition to immunotherapy, TRM cells could be targeted to enhance the efficacy of other immune interventions, such as vaccination or adoptive cell transfer. Indeed, in both settings, the induction of TRM cells enhanced their efficacy, at least in mice [17, 20]. Therefore, improving our understanding of the biology of TRM cells, including how they are generated and maintained in different human tissues, to what extent they participate in immune responses both in health (vaccination) and in disease (anti-tumor or anti-pathogen responses), and why they become dysfunctional in the tumor microenvironment, will contribute to the development of novel targeted immunotherapies.
References
Ganusov VV, De Boer RJ (2007) Do most lymphocytes in humans really reside in the gut? Trends Immunol 28(12):514–518
Masopust D, Vezys V, Marzo AL, Lefrancois L (2001) Preferential localization of effector memory cells in nonlymphoid tissue. Science 291(5512):2413–2417
Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR (2009) Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol 10(5):524–530
Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, Wang J, Casey KA, Barber DL, Kawamura KS, Fraser KA, Webby RJ, Brinkmann V, Butcher EC, Newell KA, Ahmed R (2010) Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med 207(3):553–564
Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, Kupper TS (2012) Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature 483(7388):227–231
M.E. Snyder, M.O. Finlayson, T.J. Connors, P. Dogra, T. Senda, E. Bush, D. Carpenter, C. Marboe, L. Benvenuto, L. Shah, H. Robbins, J.L. Hook, M. Sykes, F. D'Ovidio, M. Bacchetta, J.R. Sonett, D.J. Lederer, S. Arcasoy, P.A. Sims, D.L. Farber, Generation and persistence of human tissue-resident memory T cells in lung transplantation, Sci Immunol 4(33) (2019).
Bartolome-Casado R, Landsverk OJB, Chauhan SK, Richter L, Phung D, Greiff V, Risnes LF, Yao Y, Neumann RS, Yaqub S, Oyen O, Horneland R, Aandahl EM, Paulsen V, Sollid LM, Qiao SW, Baekkevold ES, Jahnsen FL (2019) Resident memory CD8 T cells persist for years in human small intestine. J Exp Med 216(10):2412–2426
Bartolome-Casado R, Landsverk OJB, Chauhan SK, Saetre F, Hagen KT, Yaqub S, Oyen O, Horneland R, Aandahl EM, Aabakken L, Baekkevold ES, Jahnsen FL (2021) CD4(+) T cells persist for years in the human small intestine and display a TH1 cytokine profile. Mucosal Immunol 14(2):402–410
L.J. Pallett, A.R. Burton, O.E. Amin, S. Rodriguez-Tajes, A.A. Patel, N. Zakeri, A. Jeffery-Smith, L. Swadling, N.M. Schmidt, A. Baiges, A. Gander, D. Yu, D. Nasralla, F. Froghi, S. Iype, B.R. Davidson, D. Thorburn, S. Yona, X. Forns, M.K. Maini, Longevity and replenishment of human liver-resident memory T cells and mononuclear phagocytes, J Exp Med 217(9) (2020).
B.G. Wiggins, L.J. Pallett, X. Li, S.P. Davies, O.E. Amin, U.S. Gill, S. Kucykowicz, A.M. Patel, K. Aliazis, Y.S. Liu, G.M. Reynolds, B.R. Davidson, A. Gander, T.V. Luong, G.M. Hirschfield, P.T.F. Kennedy, Y. Huang, M.K. Maini, Z. Stamataki, The human liver microenvironment shapes the homing and function of CD4(+) T-cell populations, Gut (2021).
Mueller SN, Mackay LK (2016) Tissue-resident memory T cells: local specialists in immune defence. Nat Rev Immunol 16(2):79–89
Dijkgraaf FE, Matos TR, Hoogenboezem M, Toebes M, Vredevoogd DW, Mertz M, van den Broek B, Song JY, Teunissen MBM, Luiten RM, Beltman JB, Schumacher TN (2019) Tissue patrol by resident memory CD8(+) T cells in human skin. Nat Immunol 20(6):756–764
J.M. Schenkel, K.A. Fraser, L.K. Beura, K.E. Pauken, V. Vezys, D. Masopust, 2014 T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses, Science 346(6205) 98–101
S. Ariotti, M.A. Hogenbirk, F.E. Dijkgraaf, L.L. Visser, M.E. Hoekstra, J.Y. Song, H. Jacobs, J.B. Haanen, T.N. Schumacher, 2014 T cell memory. Skin-resident memory CD8(+) T cells trigger a state of tissue-wide pathogen alert, Science 346(6205) 101–5.
Menares E, Galvez-Cancino F, Caceres-Morgado P, Ghorani E, Lopez E, Diaz X, Saavedra-Almarza J, Figueroa DA, Roa E, Quezada SA, Lladser A (2019) Tissue-resident memory CD8(+) T cells amplify anti-tumor immunity by triggering antigen spreading through dendritic cells. Nat Commun 10(1):4401
Eberhardt CS, Kissick HT, Patel MR, Cardenas MA, Prokhnevska N, Obeng RC, Nasti TH, Griffith CC, Im SJ, Wang X, Shin DM, Carrington M, Chen ZG, Sidney J, Sette A, Saba NF, Wieland A, Ahmed R (2021) Functional HPV-specific PD-1(+) stem-like CD8 T cells in head and neck cancer. Nature 597(7875):279–284
Nizard M, Roussel H, Diniz MO, Karaki S, Tran T, Voron T, Dransart E, Sandoval F, Riquet M, Rance B, Marcheteau E, Fabre E, Mandavit M, Terme M, Blanc C, Escudie JB, Gibault L, Barthes FLP, Granier C, Ferreira LCS, Badoual C, Johannes L, Tartour E (2017) Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat Commun 8:15221
B.T. Malik, K.T. Byrne, J.L. Vella, P. Zhang, T.B. Shabaneh, S.M. Steinberg, A.K. Molodtsov, J.S. Bowers, C.V. Angeles, C.M. Paulos, Y.H. Huang, M.J. Turk, Resident memory T cells in the skin mediate durable immunity to melanoma, Sci Immunol 2(10) (2017).
Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, Wang D, Getzler AJ, Nguyen T, Crotty S, Wang W, Pipkin ME, Goldrath AW (2017) Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature 552(7684):253–257
Enamorado M, Iborra S, Priego E, Cueto FJ, Quintana JA, Martinez-Cano S, Mejias-Perez E, Esteban M, Melero I, Hidalgo A, Sancho D (2017) Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8(+) T cells. Nat Commun 8:16073
K. Okla, D.L. Farber, W. Zou, 2021 Tissue-resident memory T cells in tumor immunity and immunotherapy, J Exp Med 218(4)
Gallatin WM, Weissman IL, Butcher EC (1983) A cell-surface molecule involved in organ-specific homing of lymphocytes. Nature 304(5921):30–34
Forster R, Schubel A, Breitfeld D, Kremmer E, Renner-Muller I, Wolf E, Lipp M (1999) CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell 99(1):23–33
Fuhlbrigge RC, Kieffer JD, Armerding D, Kupper TS (1997) Cutaneous lymphocyte antigen is a specialized form of PSGL-1 expressed on skin-homing T cells. Nature 389(6654):978–981
Berlin C, Berg EL, Briskin MJ, Andrew DP, Kilshaw PJ, Holzmann B, Weissman IL, Hamann A, Butcher EC (1993) Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell 74(1):185–195
Mora JR, Bono MR, Manjunath N, Weninger W, Cavanagh LL, Rosemblatt M, Von Andrian UH (2003) Selective imprinting of gut-homing T cells by Peyer’s patch dendritic cells. Nature 424(6944):88–93
Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A (1999) Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401(6754):708–712
Reinhardt RL, Khoruts A, Merica R, Zell T, Jenkins MK (2001) Visualizing the generation of memory CD4 T cells in the whole body. Nature 410(6824):101–105
von Andrian UH, Mackay CR (2000) T-cell function and migration Two sides of the same coin. N Engl J Med 343(14):1020–1034
Minutello MA, Pileri P, Unutmaz D, Censini S, Kuo G, Houghton M, Brunetto MR, Bonino F, Abrignani S (1993) Compartmentalization of T lymphocytes to the site of disease: intrahepatic CD4+ T cells specific for the protein NS4 of hepatitis C virus in patients with chronic hepatitis C. J Exp Med 178(1):17–25
Wong MT, Ong DE, Lim FS, Teng KW, McGovern N, Narayanan S, Ho WQ, Cerny D, Tan HK, Anicete R, Tan BK, Lim TK, Chan CY, Cheow PC, Lee SY, Takano A, Tan EH, Tam JK, Tan EY, Chan JK, Fink K, Bertoletti A, Ginhoux F, Curotto de Lafaille MA, Newell EW (2016) A high-dimensional atlas of human T cell diversity reveals tissue-specific trafficking and cytokine signatures. Immunity 45(2):442–456
Thome JJ, Yudanin N, Ohmura Y, Kubota M, Grinshpun B, Sathaliyawala T, Kato T, Lerner H, Shen Y, Farber DL (2014) Spatial map of human T cell compartmentalization and maintenance over decades of life. Cell 159(4):814–828
Miron M, Meng W, Rosenfeld AM, Dvorkin S, Poon MML, Lam N, Kumar BV, Louzoun Y, Luning Prak ET, Farber DL (2021) Maintenance of the human memory T cell repertoire by subset and tissue site. Genome Med 13(1):100
Kaech SM, Wherry EJ, Ahmed R (2002) Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol 2(4):251–262
Farber DL, Yudanin NA, Restifo NP (2014) Human memory T cells: generation, compartmentalization and homeostasis. Nat Rev Immunol 14(1):24–35
Enamorado M, Khouili SC, Iborra S, Sancho D (2018) Genealogy, dendritic cell priming, and differentiation of tissue-resident memory CD8(+) T cells. Front Immunol 9:1751
I. Raphael, R.R. Joern, T.G. Forsthuber, 2020 Memory CD4(+) T cells in immunity and autoimmune diseases, Cells 9(3)
Frost EL, Kersh AE, Evavold BD, Lukacher AE (2015) Cutting edge: Resident memory CD8 T cells express high-affinity TCRs. J Immunol 195(8):3520–3524
J.P. Snook, C. Kim, M.A. Williams, 2018 TCR signal strength controls the differentiation of CD4(+) effector and memory T cells, Sci Immunol 3(25)
Maru S, Jin G, Schell TD, Lukacher AE (2017) TCR stimulation strength is inversely associated with establishment of functional brain-resident memory CD8 T cells during persistent viral infection. PLoS Pathog 13(4):e1006318
Fiege JK, Stone IA, Fay EJ, Markman MW, Wijeyesinghe S, Macchietto MG, Shen S, Masopust D, Langlois RA (2019) The impact of TCR signal strength on resident memory T cell formation during influenza virus infection. J Immunol 203(4):936–945
Sanecka A, Yoshida N, Kolawole EM, Patel H, Evavold BD, Frickel EM (2018) T cell receptor-major histocompatibility complex interaction strength defines trafficking and CD103(+) memory status of CD8 T cells in the brain. Front Immunol 9:1290
Solouki S, Huang W, Elmore J, Limper C, Huang F, August A (2020) TCR signal strength and antigen affinity regulate CD8(+) memory T cells. J Immunol 205(5):1217–1227
L. Kok, F.E. Dijkgraaf, J. Urbanus, K. Bresser, D.W. Vredevoogd, R.F. Cardoso, L. Perie, J.B. Beltman, T.N. Schumacher, 2020 A committed tissue-resident memory T cell precursor within the circulating CD8+ effector T cell pool. J Exp Med 217(10)
V. Mani, S.K. Bromley, T. Aijo, R. Mora-Buch, E. Carrizosa, R.D. Warner, M. Hamze, D.R. Sen, A.Y. Chasse, A. Lorant, J.W. Griffith, R.A. Rahimi, C.P. McEntee, K.L. Jeffrey, F. Marangoni, M.A. Travis, A. Lacy-Hulbert, A.D. Luster, T.R. Mempel, 2019 Migratory DCs activate TGF-beta to precondition naive CD8(+) T cells for tissue-resident memory fate, Science 366(6462)
Mikhak Z, Strassner JP, Luster AD (2013) Lung dendritic cells imprint T cell lung homing and promote lung immunity through the chemokine receptor CCR4. J Exp Med 210(9):1855–1869
Campbell DJ, Butcher EC (2002) Rapid acquisition of tissue-specific homing phenotypes by CD4(+) T cells activated in cutaneous or mucosal lymphoid tissues. J Exp Med 195(1):135–141
Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY (2004) Retinoic acid imprints gut-homing specificity on T cells. Immunity 21(4):527–538
Bakdash G, Vogelpoel LT, van Capel TM, Kapsenberg ML, de Jong EC (2015) Retinoic acid primes human dendritic cells to induce gut-homing, IL-10-producing regulatory T cells. Mucosal Immunol 8(2):265–278
Sigmundsdottir H, Pan J, Debes GF, Alt C, Habtezion A, Soler D, Butcher EC (2007) DCs metabolize sunlight-induced vitamin D3 to ‘program’ T cell attraction to the epidermal chemokine CCL27. Nat Immunol 8(3):285–293
M.J. O'Melia, N.A. Rohner, M.P. Manspeaker, D.M. Francis, H.T. Kissick, S.N. Thomas, 2020 Quality of CD8(+) T cell immunity evoked in lymph nodes is compartmentalized by route of antigen transport and functional in tumor context, Sci Adv 6(50)
Wu TC, Xu K, Banchereau R, Marches F, Yu CI, Martinek J, Anguiano E, Pedroza-Gonzalez A, Snipes GJ, O’Shaughnessy J, Nishimura S, Liu YJ, Pascual V, Banchereau J, Oh S, Palucka K (2014) Reprogramming tumor-infiltrating dendritic cells for CD103+ CD8+ mucosal T-cell differentiation and breast cancer rejection. Cancer Immunol Res 2(5):487–500
P. Bourdely, G. Anselmi, K. Vaivode, R.N. Ramos, Y. Missolo-Koussou, S. Hidalgo, J. Tosselo, N. Nunez, W. Richer, A. Vincent-Salomon, A. Saxena, K. Wood, A. Lladser, E. Piaggio, J. Helft, P. Guermonprez, 2020 Transcriptional and functional analysis of CD1c(+) human dendritic cells identifies a CD163(+) subset priming CD8(+)CD103(+) T cells. Immunity 53(2) 335–352 e8.
K.D. Zens, J.K. Chen, D.L. Farber, 2016 Vaccine-generated lung tissue-resident memory T cells provide heterosubtypic protection to influenza infection, JCI Insight 1(10)
Iijima N, Iwasaki A (2014) T cell memory A local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science 346(6205):93–8
Collins N, Jiang X, Zaid A, Macleod BL, Li J, Park CO, Haque A, Bedoui S, Heath WR, Mueller SN, Kupper TS, Gebhardt T, Carbone FR (2016) Skin CD4(+) memory T cells exhibit combined cluster-mediated retention and equilibration with the circulation. Nat Commun 7:11514
Nakanishi Y, Lu B, Gerard C, Iwasaki A (2009) CD8(+) T lymphocyte mobilization to virus-infected tissue requires CD4(+) T-cell help. Nature 462(7272):510–513
Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T, Tscharke DC, Heath WR, Inouye M, Carbone FR, Gebhardt T (2013) The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol 14(12):1294–1301
Agrewala JN, Brown DM, Lepak NM, Duso D, Huston G, Swain SL (2007) Unique ability of activated CD4+ T cells but not rested effectors to migrate to non-lymphoid sites in the absence of inflammation. J Biol Chem 282(9):6106–6115
Sheridan BS, Pham QM, Lee YT, Cauley LS, Puddington L, Lefrancois L (2014) Oral infection drives a distinct population of intestinal resident memory CD8(+) T cells with enhanced protective function. Immunity 40(5):747–757
D. Herndler-Brandstetter, H. Ishigame, R. Shinnakasu, V. Plajer, C. Stecher, J. Zhao, M. Lietzenmayer, L. Kroehling, A. Takumi, K. Kometani, T. Inoue, Y. Kluger, S.M. Kaech, T. Kurosaki, T. Okada, R.A. Flavell, KLRG1(+) effector CD8(+) T cells lose KLRG1, differentiate into all memory T cell lineages, and convey enhanced protective immunity, Immunity 48(4) (2018) 716–729 e8.
Mackay LK, Minnich M, Kragten NA, Liao Y, Nota B, Seillet C, Zaid A, Man K, Preston S, Freestone D, Braun A, Wynne-Jones E, Behr FM, Stark R, Pellicci DG, Godfrey DI, Belz GT, Pellegrini M, Gebhardt T, Busslinger M, Shi W, Carbone FR, van Lier RA, Kallies A, van Gisbergen KP (2016) Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 352(6284):459–463
J.J. Milner, C. Toma, Z. He, N.S. Kurd, Q.P. Nguyen, B. McDonald, L. Quezada, C.E. Widjaja, D.A. Witherden, J.T. Crowl, L.A. Shaw, G.W. Yeo, J.T. Chang, K.D. Omilusik, A.W. Goldrath, 2020 Heterogenous populations of tissue-resident CD8(+) T cells are generated in response to infection and malignancy. Immunity 52(5) 808–824 e7
Hombrink P, Helbig C, Backer RA, Piet B, Oja AE, Stark R, Brasser G, Jongejan A, Jonkers RE, Nota B, Basak O, Clevers HC, Moerland PD, Amsen D, van Lier RA (2016) Programs for the persistence, vigilance and control of human CD8(+) lung-resident memory T cells. Nat Immunol 17(12):1467–1478
Oja AE, Piet B, Helbig C, Stark R, van der Zwan D, Blaauwgeers H, Remmerswaal EBM, Amsen D, Jonkers RE, Moerland PD, Nolte MA, van Lier RAW, Hombrink P (2018) Trigger-happy resident memory CD4(+) T cells inhabit the human lungs. Mucosal Immunol 11(3):654–667
J. Strobl, R.V. Pandey, T. Krausgruber, N. Bayer, L. Kleissl, B. Reininger, P. Vieyra-Garcia, P. Wolf, M.M. Jentus, M. Mitterbauer, P. Wohlfarth, W. Rabitsch, G. Stingl, C. Bock, G. Stary, 2020 Long-term skin-resident memory T cells proliferate in situ and are involved in human graft-versus-host disease. Sci Transl Med 12(570)
Mackay LK, Braun A, Macleod BL, Collins N, Tebartz C, Bedoui S, Carbone FR, Gebhardt T (2015) Cutting edge: CD69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J Immunol 194(5):2059–2063
Bankovich AJ, Shiow LR, Cyster JG (2010) CD69 suppresses sphingosine 1-phosophate receptor-1 (S1P1) function through interaction with membrane helix 4. J Biol Chem 285(29):22328–22337
Rivera J, Proia RL, Olivera A (2008) The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat Rev Immunol 8(10):753–763
Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, Jameson SC (2013) Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol 14(12):1285–1293
Shiow LR, Rosen DB, Brdickova N, Xu Y, An J, Lanier LL, Cyster JG, Matloubian M (2006) CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 440(7083):540–544
Cibrian D, Sanchez-Madrid F (2017) CD69: from activation marker to metabolic gatekeeper. Eur J Immunol 47(6):946–953
Baeyens AAL, Schwab SR (2020) Finding a way out: S1P signaling and immune cell migration. Annu Rev Immunol 38:759–784
M. Evrard, E. Wynne-Jones, C. Peng, Y. Kato, S.N. Christo, R. Fonseca, S.L. Park, T.N. Burn, M. Osman, S. Devi, J. Chun, S.N. Mueller, G. Kannourakis, S.P. Berzins, D.G. Pellicci, W.R. Heath, S.C. Jameson, L.K. Mackay, Sphingosine 1-phosphate receptor 5 (S1PR5) regulates the peripheral retention of tissue-resident lymphocytes, J Exp Med 219(1) (2022).
El-Asady R, Yuan R, Liu K, Wang D, Gress RE, Lucas PJ, Drachenberg CB, Hadley GA (2005) TGF-{beta}-dependent CD103 expression by CD8(+) T cells promotes selective destruction of the host intestinal epithelium during graft-versus-host disease. J Exp Med 201(10):1647–1657
Bromley SK, Akbaba H, Mani V, Mora-Buch R, Chasse AY, Sama A, Luster AD (2020) CD49a regulates cutaneous resident memory CD8(+) T cell persistence and response. Cell Rep 32(9):108085
Franciszkiewicz K, Le Floc’h A, Boutet M, Vergnon I, Schmitt A, Mami-Chouaib F (2013) CD103 or LFA-1 engagement at the immune synapse between cytotoxic T cells and tumor cells promotes maturation and regulates T-cell effector functions. Cancer Res 73(2):617–628
Meharra EJ, Schon M, Hassett D, Parker C, Havran W, Gardner H (2000) Reduced gut intraepithelial lymphocytes in VLA1 null mice. Cell Immunol 201(1):1–5
Conrad C, Boyman O, Tonel G, Tun-Kyi A, Laggner U, de Fougerolles A, Kotelianski V, Gardner H, Nestle FO (2007) Alpha1beta1 integrin is crucial for accumulation of epidermal T cells and the development of psoriasis. Nat Med 13(7):836–842
Kutlesa S, Wessels JT, Speiser A, Steiert I, Muller CA, Klein G (2002) E-cadherin-mediated interactions of thymic epithelial cells with CD103+ thymocytes lead to enhanced thymocyte cell proliferation. J Cell Sci 115(Pt 23):4505–4515
Richter MV, Topham DJ (2007) The alpha1beta1 integrin and TNF receptor II protect airway CD8+ effector T cells from apoptosis during influenza infection. J Immunol 179(8):5054–5063
Ray SJ, Franki SN, Pierce RH, Dimitrova S, Koteliansky V, Sprague AG, Doherty PC, de Fougerolles AR, Topham DJ (2004) The collagen binding alpha1beta1 integrin VLA-1 regulates CD8 T cell-mediated immune protection against heterologous influenza infection. Immunity 20(2):167–179
Heydtmann M, Lalor PF, Eksteen JA, Hubscher SG, Briskin M, Adams DH (2005) CXC chemokine ligand 16 promotes integrin-mediated adhesion of liver-infiltrating lymphocytes to cholangiocytes and hepatocytes within the inflamed human liver. J Immunol 174(2):1055–1062
Tse SW, Radtke AJ, Espinosa DA, Cockburn IA, Zavala F (2014) The chemokine receptor CXCR6 is required for the maintenance of liver memory CD8(+) T cells specific for infectious pathogens. J Infect Dis 210(9):1508–1516
Wein AN, McMaster SR, Takamura S, Dunbar PR, Cartwright EK, Hayward SL, McManus DT, Shimaoka T, Ueha S, Tsukui T, Masumoto T, Kurachi M, Matsushima K, Kohlmeier JE (2019) CXCR6 regulates localization of tissue-resident memory CD8 T cells to the airways. J Exp Med 216(12):2748–2762
Huang D, Chen X, Zeng X, Lao L, Li J, Xing Y, Lu Y, Ouyang Q, Chen J, Yang L, Su F, Yao H, Liu Q, Su S, Song E (2021) Targeting regulator of G protein signaling 1 in tumor-specific T cells enhances their trafficking to breast cancer. Nat Immunol 22(7):865–879
Lammermann T, Kastenmuller W (2019) Concepts of GPCR-controlled navigation in the immune system. Immunol Rev 289(1):205–231
Gibbons DL, Abeler-Dorner L, Raine T, Hwang IY, Jandke A, Wencker M, Deban L, Rudd CE, Irving PM, Kehrl JH, Hayday AC (2011) Cutting edge: Regulator of G protein signaling-1 selectively regulates gut T cell trafficking and colitic potential. J Immunol 187(5):2067–2071
Khan TN, Mooster JL, Kilgore AM, Osborn JF, Nolz JC (2016) Local antigen in nonlymphoid tissue promotes resident memory CD8+ T cell formation during viral infection. J Exp Med 213(6):951–966
McMaster SR, Wein AN, Dunbar PR, Hayward SL, Cartwright EK, Denning TL, Kohlmeier JE (2018) Pulmonary antigen encounter regulates the establishment of tissue-resident CD8 memory T cells in the lung airways and parenchyma. Mucosal Immunol 11(4):1071–1078
Kok L, Masopust D, Schumacher TN (2022) The precursors of CD8+ tissue resident memory T cells: from lymphoid organs to infected tissues. Nat Rev Immunol 22:283–293
R. Watanabe, A. Gehad, C. Yang, L.L. Scott, J.E. Teague, C. Schlapbach, C.P. Elco, V. Huang, T.R. Matos, T.S. Kupper, R.A. Clark, Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells, Sci Transl Med 7(279) (2015) 279ra39.
S. Cheuk, H. Schlums, I. Gallais Serezal, E. Martini, S.C. Chiang, N. Marquardt, A. Gibbs, E. Detlofsson, A. Introini, M. Forkel, C. Hoog, A. Tjernlund, J. Michaelsson, L. Folkersen, J. Mjosberg, L. Blomqvist, M. Ehrstrom, M. Stahle, Y.T. Bryceson, L. Eidsmo, 2017 CD49a expression defines tissue-resident CD8(+) T cells poised for cytotoxic function in human skin, Immunity 46(2) 287–300.
Beura LK, Fares-Frederickson NJ, Steinert EM, Scott MC, Thompson EA, Fraser KA, Schenkel JM, Vezys V, Masopust D (2019) CD4(+) resident memory T cells dominate immunosurveillance and orchestrate local recall responses. J Exp Med 216(5):1214–1229
FitzPatrick MEB, Provine NM, Garner LC, Powell K, Amini A, Irwin SL, Ferry H, Ambrose T, Friend P, Vrakas G, Reddy S, Soilleux E, Klenerman P, Allan PJ (2021) Human intestinal tissue-resident memory T cells comprise transcriptionally and functionally distinct subsets. Cell Rep 34(3):108661
Fonseca R, Beura LK, Quarnstrom CF, Ghoneim HE, Fan Y, Zebley CC, Scott MC, Fares-Frederickson NJ, Wijeyesinghe S, Thompson EA, Borges da Silva H, Vezys V, Youngblood B, Masopust D (2020) Developmental plasticity allows outside-in immune responses by resident memory T cells. Nat Immunol 21(4):412–421
Mami-Chouaib F, Blanc C, Corgnac S, Hans S, Malenica I, Granier C, Tihy I, Tartour E (2018) Resident memory T cells, critical components in tumor immunology. J Immunother Cancer 6(1):87
Amsen D, van Gisbergen K, Hombrink P, van Lier RAW (2018) Tissue-resident memory T cells at the center of immunity to solid tumors. Nat Immunol 19(6):538–546
Park SL, Buzzai A, Rautela J, Hor JL, Hochheiser K, Effern M, McBain N, Wagner T, Edwards J, McConville R, Wilmott JS, Scolyer RA, Tuting T, Palendira U, Gyorki D, Mueller SN, Huntington ND, Bedoui S, Holzel M, Mackay LK, Waithman J, Gebhardt T (2019) Tissue-resident memory CD8(+) T cells promote melanoma-immune equilibrium in skin. Nature 565(7739):366–371
Fucikova J, Kepp O, Kasikova L, Petroni G, Yamazaki T, Liu P, Zhao L, Spisek R, Kroemer G, Galluzzi L (2020) Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis 11(11):1013
Wang ZQ, Milne K, Derocher H, Webb JR, Nelson BH, Watson PH (2016) CD103 and intratumoral immune response in breast cancer. Clin Cancer Res 22(24):6290–6297
C.A. Egelston, C. Avalos, T.Y. Tu, A. Rosario, R. Wang, S. Solomon, G. Srinivasan, M.S. Nelson, Y. Huang, M.H. Lim, D.L. Simons, T.F. He, J.H. Yim, L. Kruper, J. Mortimer, S. Yost, W. Guo, C. Ruel, P.H. Frankel, Y. Yuan, P.P. Lee, 2019 Resident memory CD8+ T cells within cancer islands mediate survival in breast cancer patients, JCI Insight 4(19)
Djenidi F, Adam J, Goubar A, Durgeau A, Meurice G, de Montpreville V, Validire P, Besse B, Mami-Chouaib F (2015) CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J Immunol 194(7):3475–3486
Koh J, Kim S, Kim MY, Go H, Jeon YK, Chung DH (2017) Prognostic implications of intratumoral CD103+ tumor-infiltrating lymphocytes in pulmonary squamous cell carcinoma. Oncotarget 8(8):13762–13769
Ganesan AP, Clarke J, Wood O, Garrido-Martin EM, Chee SJ, Mellows T, Samaniego-Castruita D, Singh D, Seumois G, Alzetani A, Woo E, Friedmann PS, King EV, Thomas GJ, Sanchez-Elsner T, Vijayanand P, Ottensmeier CH (2017) Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat Immunol 18(8):940–950
Hu W, Sun R, Chen L, Zheng X, Jiang J (2019) Prognostic significance of resident CD103(+)CD8(+)T cells in human colorectal cancer tissues. Acta Histochem 121(5):657–663
T. Murray, S.A. Fuertes Marraco, P. Baumgaertner, N. Bordry, L. Cagnon, A. Donda, P. Romero, G. Verdeil, D.E. Speiser, 2016 Very late antigen-1 marks functional tumor-resident CD8 T cells and correlates with survival of melanoma patients, Front Immunol 7 573
Edwards J, Wilmott JS, Madore J, Gide TN, Quek C, Tasker A, Ferguson A, Chen J, Hewavisenti R, Hersey P, Gebhardt T, Weninger W, Britton WJ, Saw RPM, Thompson JF, Menzies AM, Long GV, Scolyer RA, Palendira U (2018) CD103(+) tumor-resident CD8(+) T cells are associated with improved survival in immunotherapy-naive melanoma patients and expand significantly during anti-PD-1 treatment. Clin Cancer Res 24(13):3036–3045
Duhen T, Duhen R, Montler R, Moses J, Moudgil T, de Miranda NF, Goodall CP, Blair TC, Fox BA, McDermott JE, Chang SC, Grunkemeier G, Leidner R, Bell RB, Weinberg AD (2018) Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat Commun 9(1):2724
Han L, Gao QL, Zhou XM, Shi C, Chen GY, Song YP, Yao YJ, Zhao YM, Wen XY, Liu SL, Qi YM, Gao YF (2020) Characterization of CD103(+) CD8(+) tissue-resident T cells in esophageal squamous cell carcinoma: may be tumor reactive and resurrected by anti-PD-1 blockade. Cancer Immunol Immunother 69(8):1493–1504
Lin R, Zhang H, Yuan Y, He Q, Zhou J, Li S, Sun Y, Li DY, Qiu HB, Wang W, Zhuang Z, Chen B, Huang Y, Liu C, Wang Y, Cai S, Ke Z, He W (2020) Fatty acid oxidation controls CD8(+) tissue-resident memory T-cell survival in gastric adenocarcinoma. Cancer Immunol Res 8(4):479–492
Lim CJ, Lee YH, Pan L, Lai L, Chua C, Wasser M, Lim TKH, Yeong J, Toh HC, Lee SY, Chan CY, Goh BK, Chung A, Heikenwalder M, Ng IO, Chow P, Albani S, Chew V (2019) Multidimensional analyses reveal distinct immune microenvironment in hepatitis B virus-related hepatocellular carcinoma. Gut 68(5):916–927
Webb JR, Milne K, Watson P, Deleeuw RJ, Nelson BH (2014) Tumor-infiltrating lymphocytes expressing the tissue resident memory marker CD103 are associated with increased survival in high-grade serous ovarian cancer. Clin Cancer Res 20(2):434–444
Bosmuller HC, Wagner P, Peper JK, Schuster H, Pham DL, Greif K, Beschorner C, Rammensee HG, Stevanovic S, Fend F, Staebler A (2016) Combined immunoscore of CD103 and CD3 identifies long-term survivors in high-grade serous ovarian cancer. Int J Gynecol Cancer 26(4):671–679
Komdeur FL, Wouters MC, Workel HH, Tijans AM, Terwindt AL, Brunekreeft KL, Plat A, Klip HG, Eggink FA, Leffers N, Helfrich W, Samplonius DF, Bremer E, Wisman GB, Daemen T, Duiker EW, Hollema H, Nijman HW, de Bruyn M (2016) CD103+ intraepithelial T cells in high-grade serous ovarian cancer are phenotypically diverse TCRalphabeta+ CD8alphabeta+ T cells that can be targeted for cancer immunotherapy. Oncotarget 7(46):75130–75144
Komdeur FL, Prins TM, van de Wall S, Plat A, Wisman GBA, Hollema H, Daemen T, Church DN, de Bruyn M, Nijman HW (2017) CD103+ tumor-infiltrating lymphocytes are tumor-reactive intraepithelial CD8+ T cells associated with prognostic benefit and therapy response in cervical cancer. Oncoimmunology 6(9):e1338230
Workel HH, Komdeur FL, Wouters MC, Plat A, Klip HG, Eggink FA, Wisman GB, Arts HJ, Oonk MH, Mourits MJ, Yigit R, Versluis M, Duiker EW, Hollema H, de Bruyn M, Nijman HW (2016) CD103 defines intraepithelial CD8+ PD1+ tumour-infiltrating lymphocytes of prognostic significance in endometrial adenocarcinoma. Eur J Cancer 60:1–11
Wang B, Wu S, Zeng H, Liu Z, Dong W, He W, Chen X, Dong X, Zheng L, Lin T, Huang J (2015) CD103+ tumor infiltrating lymphocytes predict a favorable prognosis in urothelial cell carcinoma of the bladder. J Urol 194(2):556–562
Le Floc’h A, Jalil A, Vergnon I, Le Maux Chansac B, Lazar V, Bismuth G, Chouaib S, Mami-Chouaib F (2007) Alpha E beta 7 integrin interaction with E-cadherin promotes antitumor CTL activity by triggering lytic granule polarization and exocytosis. J Exp Med 204(3):559–70
Le Floc’h A, Jalil A, Franciszkiewicz K, Validire P, Vergnon I, Mami-Chouaib F (2011) Minimal engagement of CD103 on cytotoxic T lymphocytes with an E-cadherin-Fc molecule triggers lytic granule polarization via a phospholipase Cgamma-dependent pathway. Cancer Res 71(2):328–338
E.C. Reilly, K. Lambert Emo, P.M. Buckley, N.S. Reilly, I. Smith, F.A. Chaves, H. Yang, P.W. Oakes, D.J. Topham, TRM integrins CD103 and CD49a differentially support adherence and motility after resolution of influenza virus infection, Proc Natl Acad Sci U S A 117(22) (2020) 12306–12314.
Schenkel JM, Fraser KA, Vezys V, Masopust D (2013) Sensing and alarm function of resident memory CD8(+) T cells. Nat Immunol 14(5):509–513
McMaster SR, Wilson JJ, Wang H, Kohlmeier JE (2015) Airway-resident memory CD8 T cells provide antigen-specific protection against respiratory virus challenge through rapid IFN-gamma production. J Immunol 195(1):203–209
J.S. Low, Y. Farsakoglu, M.C. Amezcua Vesely, E. Sefik, J.B. Kelly, C.C.D. Harman, R. Jackson, J.A. Shyer, X. Jiang, L.S. Cauley, R.A. Flavell, S.M. Kaech, 2020 Tissue-resident memory T cell reactivation by diverse antigen-presenting cells imparts distinct functional responses. J Exp Med 217(8)
Sathaliyawala T, Kubota M, Yudanin N, Turner D, Camp P, Thome JJ, Bickham KL, Lerner H, Goldstein M, Sykes M, Kato T, Farber DL (2013) Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity 38(1):187–197
Sanchez Rodriguez R, Pauli ML, Neuhaus IM, Yu SS, Arron ST, Harris HW, Yang SH, Anthony BA, Sverdrup FM, Krow-Lucal E, MacKenzie TC, Johnson DS, Meyer EH, Lohr A, Hsu A, Koo J, Liao W, Gupta R, Debbaneh MG, Butler D, Huynh M, Levin EC, Leon A, Hoffman WY, McGrath MH, Alvarado MD, Ludwig CH, Truong HA, Maurano MM, Gratz IK, Abbas AK, Rosenblum MD (2014) Memory regulatory T cells reside in human skin. J Clin Invest 124(3):1027–36
Kleinschek MA, Boniface K, Sadekova S, Grein J, Murphy EE, Turner SP, Raskin L, Desai B, Faubion WA, de Waal Malefyt R, Pierce RH, McClanahan T, Kastelein RA (2009) Circulating and gut-resident human Th17 cells express CD161 and promote intestinal inflammation. J Exp Med 206(3):525–34
J. Strobl, L.M. Gail, L. Kleissl, R.V. Pandey, V. Smejkal, J. Huber, V. Puxkandl, L. Unterluggauer, R. Dingelmaier-Hovorka, D. Atzmuller, T. Krausgruber, C. Bock, P. Wohlfarth, W. Rabitsch, G. Stary, 2021 Human resident memory T cells exit the skin and mediate systemic Th2-driven inflammation. J Exp Med 218(11)
Y. Zhao, C. Kilian, J.E. Turner, L. Bosurgi, K. Roedl, P. Bartsch, A.C. Gnirck, F. Cortesi, C. Schultheiss, M. Hellmig, L.U.B. Enk, F. Hausmann, A. Borchers, M.N. Wong, H.J. Paust, F. Siracusa, N. Scheibel, M. Herrmann, E. Rosati, P. Bacher, D. Kylies, D. Jarczak, M. Lutgehetmann, S. Pfefferle, S. Steurer, J.S. Zur-Wiesch, V.G. Puelles, J.P. Sperhake, M.M. Addo, A.W. Lohse, M. Binder, S. Huber, T.B. Huber, S. Kluge, S. Bonn, U. Panzer, N. Gagliani, C.F. Krebs, 2021 Clonal expansion and activation of tissue-resident memory-like Th17 cells expressing GM-CSF in the lungs of severe COVID-19 patients, Sci Immunol 6(56)
Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, Jungbluth A, Gnjatic S, Thompson JA, Yee C (2008) Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med 358(25):2698–2703
Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, Parkhurst MR, Yang JC, Rosenberg SA (2014) Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344(6184):641–645
Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ, Behjati S, Velds A, Hilkmann H, Atmioui DE, Visser M, Stratton MR, Haanen JB, Spits H, van der Burg SH, Schumacher TN (2015) High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med 21(1):81–85
Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, Chen C, Olive O, Carter TA, Li S, Lieb DJ, Eisenhaure T, Gjini E, Stevens J, Lane WJ, Javeri I, Nellaiappan K, Salazar AM, Daley H, Seaman M, Buchbinder EI, Yoon CH, Harden M, Lennon N, Gabriel S, Rodig SJ, Barouch DH, Aster JC, Getz G, Wucherpfennig K, Neuberg D, Ritz J, Lander ES, Fritsch EF, Hacohen N, Wu CJ (2017) An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547(7662):217–221
Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, Oliveira G, Giobbie-Hurder A, Felt K, Gjini E, Shukla SA, Hu Z, Li L, Le PM, Allesoe RL, Richman AR, Kowalczyk MS, Abdelrahman S, Geduldig JE, Charbonneau S, Pelton K, Iorgulescu JB, Elagina L, Zhang W, Olive O, McCluskey C, Olsen LR, Stevens J, Lane WJ, Salazar AM, Daley H, Wen PY, Chiocca EA, Harden M, Lennon NJ, Gabriel S, Getz G, Lander ES, Regev A, Ritz J, Neuberg D, Rodig SJ, Ligon KL, Suva ML, Wucherpfennig KW, Hacohen N, Fritsch EF, Livak KJ, Ott PA, Wu CJ, Reardon DA (2019) Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565(7738):234–239
Alspach E, Lussier DM, Miceli AP, Kizhvatov I, DuPage M, Luoma AM, Meng W, Lichti CF, Esaulova E, Vomund AN, Runci D, Ward JP, Gubin MM, Medrano RFV, Arthur CD, White JM, Sheehan KCF, Chen A, Wucherpfennig KW, Jacks T, Unanue ER, Artyomov MN, Schreiber RD (2019) MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 574(7780):696–701
T. Ahrends, A. Spanjaard, B. Pilzecker, N. Babala, A. Bovens, Y. Xiao, H. Jacobs, J. Borst, CD4(+) T cell help confers a cytotoxic T cell effector program including coinhibitory receptor downregulation and increased tissue invasiveness, Immunity 47(5) (2017) 848–861 e5.
Hirschhorn-Cymerman D, Budhu S, Kitano S, Liu C, Zhao F, Zhong H, Lesokhin AM, Avogadri-Connors F, Yuan J, Li Y, Houghton AN, Merghoub T, Wolchok JD (2012) Induction of tumoricidal function in CD4+ T cells is associated with concomitant memory and terminally differentiated phenotype. J Exp Med 209(11):2113–2126
Khodadoust MS, Olsson N, Wagar LE, Haabeth OA, Chen B, Swaminathan K, Rawson K, Liu CL, Steiner D, Lund P, Rao S, Zhang L, Marceau C, Stehr H, Newman AM, Czerwinski DK, Carlton VE, Moorhead M, Faham M, Kohrt HE, Carette J, Green MR, Davis MM, Levy R, Elias JE, Alizadeh AA (2017) Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. Nature 543(7647):723–727
Bailey SR, Nelson MH, Majchrzak K, Bowers JS, Wyatt MM, Smith AS, Neal LR, Shirai K, Carpenito C, June CH, Zilliox MJ, Paulos CM (2017) Human CD26(high) T cells elicit tumor immunity against multiple malignancies via enhanced migration and persistence. Nat Commun 8(1):1961
Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, Johansson I, Phung B, Harbst K, Vallon-Christersson J, van Schoiack A, Lovgren K, Warren S, Jirstrom K, Olsson H, Pietras K, Ingvar C, Isaksson K, Schadendorf D, Schmidt H, Bastholt L, Carneiro A, Wargo JA, Svane IM, Jonsson G (2020) Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577(7791):561–565
Petitprez F, de Reynies A, Keung EZ, Chen TW, Sun CM, Calderaro J, Jeng YM, Hsiao LP, Lacroix L, Bougouin A, Moreira M, Lacroix G, Natario I, Adam J, Lucchesi C, Laizet YH, Toulmonde M, Burgess MA, Bolejack V, Reinke D, Wani KM, Wang WL, Lazar AJ, Roland CL, Wargo JA, Italiano A, Sautes-Fridman C, Tawbi HA, Fridman WH (2020) B cells are associated with survival and immunotherapy response in sarcoma. Nature 577(7791):556–560
I. Kryczek, E. Zhao, Y. Liu, Y. Wang, L. Vatan, W. Szeliga, J. Moyer, A. Klimczak, A. Lange, W. Zou, 2011 Human TH17 cells are long-lived effector memory cells, Sci Transl Med 3(104) 104ra100.
Kryczek I, Banerjee M, Cheng P, Vatan L, Szeliga W, Wei S, Huang E, Finlayson E, Simeone D, Welling TH, Chang A, Coukos G, Liu R, Zou W (2009) Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood 114(6):1141–1149
P. Ogongo, L.B. Tezera, A. Ardain, S. Nhamoyebonde, D. Ramsuran, A. Singh, A. Ng'oepe, F. Karim, T. Naidoo, K. Khan, K.J. Dullabh, M. Fehlings, B.H. Lee, A. Nardin, C.S. Lindestam Arlehamn, A. Sette, S.M. Behar, A.J. Steyn, R. Madansein, H.N. Kloverpris, P.T. Elkington, A. Leslie, 2021 Tissue-resident-like CD4+ T cells secreting IL-17 control Mycobacterium tuberculosis in the human lung, J Clin Invest 131(10)
M.C. Amezcua Vesely, P. Pallis, P. Bielecki, J.S. Low, J. Zhao, C.C.D. Harman, L. Kroehling, R. Jackson, W. Bailis, P. Licona-Limon, H. Xu, N. Iijima, P.S. Pillai, D.H. Kaplan, C.T. Weaver, Y. Kluger, M.S. Kowalczyk, A. Iwasaki, J.P. Pereira, E. Esplugues, N. Gagliani, R.A. Flavell, 2019 Effector TH17 cells give rise to long-lived TRM cells that are essential for an immediate response against bacterial infection, Cell 178(5) 1176–1188 e15.
Aschenbrenner D, Foglierini M, Jarrossay D, Hu D, Weiner HL, Kuchroo VK, Lanzavecchia A, Notarbartolo S, Sallusto F (2018) An immunoregulatory and tissue-residency program modulated by c-MAF in human TH17 cells. Nat Immunol 19(10):1126–1136
Oja AE, Piet B, van der Zwan D, Blaauwgeers H, Mensink M, de Kivit S, Borst J, Nolte MA, van Lier RAW, Stark R, Hombrink P (2018) Functional heterogeneity of CD4(+) tumor-infiltrating lymphocytes with a resident memory phenotype in NSCLC. Front Immunol 9:2654
De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, Bonnal RJP, Provasi E, Sarnicola ML, Panzeri I, Moro M, Crosti M, Mazzara S, Vaira V, Bosari S, Palleschi A, Santambrogio L, Bovo G, Zucchini N, Totis M, Gianotti L, Cesana G, Perego RA, Maroni N, Pisani Ceretti A, Opocher E, De Francesco R, Geginat J, Stunnenberg HG, Abrignani S, Pagani M (2016) Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor-infiltrating T regulatory cells. Immunity 45(5):1135–1147
Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV, Chudakov DM, Rudensky AY (2016) Regulatory T cells exhibit distinct features in human breast cancer. Immunity 45(5):1122–1134
McCully ML, Ladell K, Andrews R, Jones RE, Miners KL, Roger L, Baird DM, Cameron MJ, Jessop ZM, Whitaker IS, Davies EL, Price DA, Moser B (2018) CCR8 expression defines tissue-resident memory T cells in human skin. J Immunol 200(5):1639–1650
Bonnal RJP, Rossetti G, Lugli E, De Simone M, Gruarin P, Brummelman J, Drufuca L, Passaro M, Bason R, Gervasoni F, Della Chiara G, D’Oria C, Martinovic M, Curti S, Ranzani V, Cordiglieri C, Alvisi G, Mazza EMC, Oliveto S, Silvestri Y, Carelli E, Mazzara S, Bosotti R, Sarnicola ML, Godano C, Bevilacqua V, Lorenzo M, Siena S, Bonoldi E, Sartore-Bianchi A, Amatu A, Veronesi G, Novellis P, Alloisio M, Giani A, Zucchini N, Opocher E, Ceretti AP, Mariani N, Biffo S, Prati D, Bardelli A, Geginat J, Lanzavecchia A, Abrignani S, Pagani M (2021) Clonally expanded EOMES(+) Tr1-like cells in primary and metastatic tumors are associated with disease progression. Nat Immunol 22(6):735–745
F. Facciotti, N. Gagliani, B. Haringer, J.S. Alfen, A. Penatti, S. Maglie, M. Paroni, A. Iseppon, M. Moro, M.C. Crosti, K. Stolzel, C. Romagnani, G. Moroni, F. Ingegnoli, S. Torretta, L. Pignataro, A. Annoni, F. Russo, M. Pagani, S. Abrignani, P. Meroni, R. Flavell, J. Geginat02016, IL-10-producing forkhead box protein 3-negative regulatory T cells inhibit B-cell responses and are involved in systemic lupus erythematosus, J Allergy Clin Immunol 137(1) 318–321 e5.
A.S. Woodward Davis, H.N. Roozen, M.J. Dufort, H.A. DeBerg, M.A. Delaney, F. Mair, J.R. Erickson, C.K. Slichter, J.D. Berkson, A.M. Klock, M. Mack, Y. Lwo, A. Ko, R.M. Brand, I. McGowan, P.S. Linsley, D.R. Dixon, M. Prlic, 2019 The human tissue-resident CCR5(+) T cell compartment maintains protective and functional properties during inflammation. Sci Transl Med 11(521)
E. Azizi, A.J. Carr, G. Plitas, A.E. Cornish, C. Konopacki, S. Prabhakaran, J. Nainys, K. Wu, V. Kiseliovas, M. Setty, K. Choi, R.M. Fromme, P. Dao, P.T. McKenney, R.C. Wasti, K. Kadaveru, L. Mazutis, A.Y. Rudensky, D. Pe'er, 2018 Single-cell map of diverse immune phenotypes in the breast tumor microenvironment, Cell 174(5) 1293–1308 e36.
P. Savas, B. Virassamy, C. Ye, A. Salim, C.P. Mintoff, F. Caramia, R. Salgado, D.J. Byrne, Z.L. Teo, S. Dushyanthen, A. Byrne, L. Wein, S.J. Luen, C. Poliness, S.S. Nightingale, A.S. Skandarajah, D.E. Gyorki, C.M. Thornton, P.A. Beavis, S.B. Fox, C. Kathleen Cuningham Foundation Consortium for Research into Familial Breast, P.K. Darcy, T.P. Speed, L.K. Mackay, P.J. Neeson, S. Loi, Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis, Nat Med 24(7) (2018) 986-993.
Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, Kang B, Liu Z, Jin L, Xing R, Gao R, Zhang L, Dong M, Hu X, Ren X, Kirchhoff D, Roider HG, Yan T, Zhang Z (2018) Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med 24(7):978–985
Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, Gao R, Kang B, Zhang Q, Huang JY, Konno H, Guo X, Ye Y, Gao S, Wang S, Hu X, Ren X, Shen Z, Ouyang W, Zhang Z (2018) Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 564(7735):268–272
C. Zheng, L. Zheng, J.K. Yoo, H. Guo, Y. Zhang, X. Guo, B. Kang, R. Hu, J.Y. Huang, Q. Zhang, Z. Liu, M. Dong, X. Hu, W. Ouyang, J. Peng, Z. Zhang, 2017 Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 69(7) 1342–1356 e16
D.Y. Oh, S.S. Kwek, S.S. Raju, T. Li, E. McCarthy, E. Chow, D. Aran, A. Ilano, C.S. Pai, C. Rancan, K. Allaire, A. Burra, Y. Sun, M.H. Spitzer, S. Mangul, S. Porten, M.V. Meng, T.W. Friedlander, C.J. Ye, L. Fong, 2020 Intratumoral CD4(+) T cells mediate anti-tumor cytotoxicity in human bladder cancer, Cell 181(7) 1612–1625 e13.
A.R. Cillo, C.H.L. Kurten, T. Tabib, Z. Qi, S. Onkar, T. Wang, A. Liu, U. Duvvuri, S. Kim, R.J. Soose, S. Oesterreich, W. Chen, R. Lafyatis, T.C. Bruno, R.L. Ferris, D.A.A. Vignali, 2020 Immune landscape of viral- and carcinogen-driven head and neck cancer, Immunity 52(1) (183–199 e9.
Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, McNamara KL, Granja JM, Sarin KY, Brown RA, Gupta RK, Curtis C, Bucktrout SL, Davis MM, Chang ALS, Chang HY (2019) Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med 25(8):1251–1259
Satpathy AT, Granja JM, Yost KE, Qi Y, Meschi F, McDermott GP, Olsen BN, Mumbach MR, Pierce SE, Corces MR, Shah P, Bell JC, Jhutty D, Nemec CM, Wang J, Wang L, Yin Y, Giresi PG, Chang ALS, Zheng GXY, Greenleaf WJ, Chang HY (2019) Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat Biotechnol 37(8):925–936
Wu TD, Madireddi S, de Almeida PE, Banchereau R, Chen YJ, Chitre AS, Chiang EY, Iftikhar H, O’Gorman WE, Au-Yeung A, Takahashi C, Goldstein LD, Poon C, Keerthivasan S, de Almeida Nagata DE, Du X, Lee HM, Banta KL, Mariathasan S, Das Thakur M, Huseni MA, Ballinger M, Estay I, Caplazi P, Modrusan Z, Delamarre L, Mellman I, Bourgon R, Grogan JL (2020) Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 579(7798):274–278
Boddupalli CS, Bar N, Kadaveru K, Krauthammer M, Pornputtapong N, Mai Z, Ariyan S, Narayan D, Kluger H, Deng Y, Verma R, Das R, Bacchiocchi A, Halaban R, Sznol M, Dhodapkar MV, Dhodapkar KM (2016) Interlesional diversity of T cell receptors in melanoma with immune checkpoints enriched in tissue-resident memory T cells. JCI Insight 1(21):e88955
Corgnac S, Malenica I, Mezquita L, Auclin E, Voilin E, Kacher J, Halse H, Grynszpan L, Signolle N, Dayris T, Leclerc M, Droin N, de Montpreville V, Mercier O, Validire P, Scoazec JY, Massard C, Chouaib S, Planchard D, Adam J, Besse B, Mami-Chouaib F (2020) CD103(+)CD8(+) TRM cells accumulate in tumors of anti-PD-1-responder lung cancer patients and are tumor-reactive lymphocytes enriched with Tc17. Cell Rep Med 1(7):100127
Ngiow SF, Young A, Jacquelot N, Yamazaki T, Enot D, Zitvogel L, Smyth MJ (2015) A threshold level of intratumor CD8+ T-cell PD1 expression dictates therapeutic response to anti-PD1. Cancer Res 75(18):3800–3811
Zhang N, Bevan MJ (2013) Transforming growth factor-beta signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity 39(4):687–696
Mackay LK, Wynne-Jones E, Freestone D, Pellicci DG, Mielke LA, Newman DM, Braun A, Masson F, Kallies A, Belz GT, Carbone FR (2015) T-box transcription factors combine with the cytokines TGF-beta and IL-15 to control tissue-resident memory T cell fate. Immunity 43(6):1101–1111
F. Hasan, Y. Chiu, R.M. Shaw, J. Wang, C. Yee, Hypoxia acts as an environmental cue for the human tissue-resident memory T cell differentiation program, JCI Insight 6(10) (2021).
M.Y. Braun, The Natural History of T Cell Metabolism, Int J Mol Sci 22(13) (2021).
Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu X, Luo C, O’Malley JT, Gehad A, Teague JE, Divito SJ, Fuhlbrigge R, Puigserver P, Krueger JG, Hotamisligil GS, Clark RA, Kupper TS (2017) Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 543(7644):252–256
Buck MD, Sowell RT, Kaech SM, Pearce EL (2017) Metabolic instruction of immunity. Cell 169(4):570–586
Y. Zhang, R. Kurupati, L. Liu, X.Y. Zhou, G. Zhang, A. Hudaihed, F. Filisio, W. Giles-Davis, X. Xu, G.C. Karakousis, L.M. Schuchter, W. Xu, R. Amaravadi, M. Xiao, N. Sadek, C. Krepler, M. Herlyn, G.J. Freeman, J.D. Rabinowitz, H.C.J. Ertl, 2017 Enhancing CD8(+) T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy, Cancer Cell 32(3) 377–391 e9.
D.C. Fuhrmann, C. Olesch, N. Kurrle, F. Schnutgen, S. Zukunft, I. Fleming, B. Brune, Chronic hypoxia enhances beta-oxidation-dependent electron transport via electron transferring flavoproteins, Cells 8(2) (2019).
R. Geiger, J.C. Rieckmann, T. Wolf, C. Basso, Y. Feng, T. Fuhrer, M. Kogadeeva, P. Picotti, F. Meissner, M. Mann, N. Zamboni, F. Sallusto, A. Lanzavecchia, 2016 L-Arginine modulates T cell metabolism and enhances survival and anti-tumor activity, Cell 167(3) 829–842 e13.
Canale FP, Basso C, Antonini G, Perotti M, Li N, Sokolovska A, Neumann J, James MJ, Geiger S, Jin W, Theurillat JP, West KA, Leventhal DS, Lora JM, Sallusto F, Geiger R (2021) Metabolic modulation of tumours with engineered bacteria for immunotherapy. Nature 598(7882):662–666
Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, Yeong JPS, Nahar R, Zhang T, Kared H, Duan K, Ang N, Poidinger M, Lee YY, Larbi A, Khng AJ, Tan E, Fu C, Mathew R, Teo M, Lim WT, Toh CK, Ong BH, Koh T, Hillmer AM, Takano A, Lim TKH, Tan EH, Zhai W, Tan DSW, Tan IB, Newell EW (2018) Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 557(7706):575–579
A. Martinez-Usatorre, S.J. Carmona, C. Godfroid, C. Yacoub Maroun, S. Labiano, P. Romero, Enhanced phenotype definition for precision isolation of precursor exhausted tumor-infiltrating CD8 T cells, Front Immunol 11 (2020) 340.
Acknowledgements
We thank S. Biffo for the discussion on T cell metabolism. Figures were created with Biorender.com.
Funding
Open access funding provided by Università degli Studi di Milano within the CRUI-CARE Agreement. S. Abrignani is supported by Fondazione AIRC under 5 per Mille 2018 (AIRC 5 × 1000 programs: project 21147 and project 21091) and Cancer Research UK Accelerator Award no. 22794.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
This article is a contribution to the special issue on: Heterogeneity of tissue-resident immunity across organs and in health and disease - Guest Editors: Federica Sallusto &; Petra Arck
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Notarbartolo, S., Abrignani, S. Human T lymphocytes at tumor sites. Semin Immunopathol 44, 883–901 (2022). https://doi.org/10.1007/s00281-022-00970-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-022-00970-4