
Abstract
Aims/hypothesis
Pancreatic beta cell dedifferentiation, transdifferentiation into other islet cells and apoptosis have been implicated in beta cell failure in type 2 diabetes, although the mechanisms are poorly defined. The endoplasmic reticulum stress response factor X-box binding protein 1 (XBP1) is a major regulator of the unfolded protein response. XBP1 expression is reduced in islets of people with type 2 diabetes, but its role in adult differentiated beta cells is unclear. Here, we assessed the effects of Xbp1 deletion in adult beta cells and tested whether XBP1-mediated unfolded protein response makes a necessary contribution to beta cell compensation in insulin resistance states.
Methods
Mice with inducible beta cell-specific Xbp1 deletion were studied under normal (chow diet) or metabolic stress (high-fat diet or obesity) conditions. Glucose tolerance, insulin secretion, islet gene expression, alpha cell mass, beta cell mass and apoptosis were assessed. Lineage tracing was used to determine beta cell fate.
Results
Deletion of Xbp1 in adult mouse beta cells led to beta cell dedifferentiation, beta-to-alpha cell transdifferentiation and increased alpha cell mass. Cell lineage-specific analyses revealed that Xbp1 deletion deactivated beta cell identity genes (insulin, Pdx1, Nkx6.1, Beta2, Foxo1) and derepressed beta cell dedifferentiation (Aldh1a3) and alpha cell (glucagon, Arx, Irx2) genes. Xbp1 deletion in beta cells of obese ob/ob or high-fat diet-fed mice triggered diabetes and worsened glucose intolerance by disrupting insulin secretory capacity. Furthermore, Xbp1 deletion increased beta cell apoptosis under metabolic stress conditions by attenuating the antioxidant response.
Conclusions/interpretation
These findings indicate that XBP1 maintains beta cell identity, represses beta-to-alpha cell transdifferentiation and is required for beta cell compensation and prevention of diabetes in insulin resistance states.
Graphical abstract

Similar content being viewed by others


Introduction
The ability of pancreatic beta cells to augment insulin production and secretion in response to increased metabolic demand is crucial for the maintenance of normoglycaemia. This adaptive response, termed beta cell compensation, is associated with both enhanced beta cell function and expansion of beta cell mass [1,2,3]. Hyperglycaemia develops when the demand for insulin exceeds the functional capacity of beta cells. Beta cell dysfunction occurs early in the aetiology of type 2 diabetes and deteriorates as the diabetic state worsens and beta cell capacity declines [4]. Deficits in adaptive beta cell mass linked with increased apoptosis are thought to contribute to the course of type 2 diabetes [5]. While much attention has focused on elucidating the mechanisms of beta cell compensation and failure in the development and progression of type 2 diabetes, they remain poorly understood.
Beta cell dedifferentiation has been proposed as an important contributor to beta cell failure in type 2 diabetes [6,7,8,9]. Evidence from animal and human studies suggests that mature beta cells lose their differentiated phenotype and cellular identity and regress to a less differentiated state and/or transdifferentiate into other islet cells in type 2 diabetes. While a major role of glucotoxicity in beta cell dedifferentiation has been established [6, 7, 10,11,12], the precise molecular mechanisms involved remain unclear.
The production of large quantities of insulin in beta cells imposes significant burden on the endoplasmic reticulum (ER) [13, 14]. Adaptation to ER stress by unfolded protein response (UPR) activation is proposed to play a protective role in increased insulin production, whereas the failure of adaptation has been implicated in beta cell dysfunction and apoptosis [15,16,17,18,19,20]. The UPR is mediated by three transmembrane stress sensor proteins: PKR-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1) and activating transcription factor 6 (ATF6). IRE1 cleaves the Xbp1 mRNA, generating a transcription factor that promotes ER biogenesis and activates the expression of ER chaperone genes that are required for the folding and trafficking of secretory proteins. X-box binding protein 1 (XBP1) is an important regulator of the ER stress response in beta cells [18, 21,22,23,24,25,26,27,28]. Decreased spliced XBP1 expression was observed in islets of type 2 diabetic patients [29] and studies adopting embryonic deletion of Xbp1 using the rat insulin promoter showed that it is required for optimal beta cell development and proinsulin processing [21]. Here, we employed inducible deletion of Xbp1 specifically in beta cells of adult mice to investigate the function of XBP1 in the adaptive response of mature beta cells to increased metabolic demand.
Methods
For detailed methods, please refer to the electronic supplementary material (ESM) Methods.
Mouse models
To enable tamoxifen-inducible beta cell-specific deletion of Xbp1, we crossed Pdx1-CreER mice [Tg(Pdx1-cre/Esr1*)#Dam/J, The Jackson Laboratory, Bar Harbour, ME, USA] with Xbp1flox mice kindly provided by L. Glimcher, Dana-Farber Cancer Institute, Harvard University, Boston, MA, USA [21]. Experimental mice received i.p. tamoxifen (Sigma-Aldrich, St Louis, MO, USA) injections (3 × 75 mg/kg body weight) at 8–10 weeks of age to generate Xbp1flox/flox Pdx1-CreER (denoted β-Xbp1−/−) and littermate control Xbp1+/+ Pdx1-CreER (β-Xbp1+/+) mice (ESM Fig. 1). Male littermates were assigned randomly to either chow or a high-fat diet.
To enable tamoxifen-inducible beta cell-specific deletion of Xbp1 in obese mice, we crossed Xbp1flox/flox Pdx1-CreER mice with ob/+ (B6.Cg-Lepob/J; The Jackson Laboratory) mice. Experimental mice received i.p. tamoxifen injections at 8–10 weeks of age to generate Xbp1flox/flox Pdx1-CreER ob/ob (β-Xbp1−/−Ob) and Xbp1flox/flox Pdx1-CreER wild-type/wild-type (β-Xbp1−/−Wt) mice and control Xbp1+/+ Pdx1-CreER ob/ob (β-Xbp1+/+Ob) and Xbp1+/+ Pdx1-CreER wild-type/wild-type (β-Xbp1+/+Wt) mice.
A model for cell type-specific lineage tracing and transcript profiling was developed by crossing Xbp1flox/flox Pdx1-CreER mice with Gt/Rosa26GFP [Gt(ROSA)26Sortm9(EGFP/Rpl10a)Amc; The Jackson Laboratory] mice. Mice received i.p. tamoxifen injections at 8–10 weeks of age to generate Xbp1flox/flox Pdx1-CreER Gt/Rosa26GFP (β-Xbp1−/−Gt) and control Xbp1+/+ Pdx1-CreER Gt/Rosa26GFP (β-Xbp1+/+Gt) mice (see text box for mouse models).
Glucose tolerance test
Glucose tolerance tests were performed after a 6 h fast, by oral gavage administration of glucose (1.5 or 3 g/kg body weight).
Islet isolation and insulin secretion assays
Islets were isolated by Liberase digestion (Roche Diagnostics, Castle Hill, Australia), gradient centrifugation (Ficoll-Paque PLUS gradient, GE Healthcare Bio-Sciences, Uppsala, Sweden) and handpicking under a stereomicroscope. To assess ex vivo insulin secretion, batches of five islets were incubated in KRB-HEPES supplemented with 0.1% BSA containing either 2 or 20 mmol/l glucose for 1 h at 37°C. Secreted insulin was determined using the Insulin Ultra-Sensitive Assay (Cisbio, Codolet, France). The islets were lysed for measurement of insulin and DNA content.
Glucolipotoxicity and antioxidant treatment ex vivo
Islets isolated from littermate Xbp1+/+ Pdx1-CreER and Xbp1flox/flox Pdx1-CreER mice were treated with 100 nmol/l 4-hydroxy tamoxifen (Sigma-Aldrich) to generate β-Xbp1+/+ and β-Xbp1−/− islets. Next, islets were treated in islet media with either 0.92% BSA (control) or 25 mmol/l glucose and 0.4 mmol/l palmitate coupled to 0.92% BSA (termed glucose + palmitate, GP) for 72 h at 37°C. For antioxidant treatment experiments, islets were co-treated with 2.5 mmol/l N-acetyl-l-cysteine (NAC) (Sigma-Aldrich). Cell death was determined in islets using the Cell Death Detection ELISAPLUS Kit (Roche Diagnostics).
Histology and immuno-morphometry
Formalin-fixed paraffin-embedded pancreases were sectioned and immunostained using the antibodies indicated (ESM Table 1). Slides were imaged using a Leica DM5500 fluorescent microscope or Leica DM6000 Power Mosaic microscope (Leica Microsystems, Wetzlar, Germany). Immunostaining was quantified using ImageJ/FIJI (ImageJ Developers; version 1.52a; https://imageJ.nih.gov/ij/; and FIJI Contributors; version 2.0.0-rc-69/1.52n; https://fiji.sc/).
Immunoblotting
Islets were analysed by immunoblotting for XBP1, 14–3-3, phosphorylated IRE1 and total IRE1 using the antibodies indicated (ESM Table 1).
RNA analysis
Real-time PCR was performed on the 7900 HT Real Time PCR System (Applied Biosystems, Foster City, CA, USA) using oligonucleotide primers (ESM Table 2). The value obtained for each specific gene product was normalised to a housekeeping gene (cyclophilin A) and expressed as a fold change of the value in control extracts.
Translating ribosome affinity purification
Translating ribosome affinity purification (TRAP) was performed based on the previously published protocol [30, 31].
Statistical analysis
All data are represented as means ± SEM. Unpaired two-tailed t test was used to compare differences between two groups. Differences between more than two groups were calculated using two-way ANOVA with Tukey’s post hoc test.
Results
Beta cell-specific deletion of Xbp1 in high-fat diet-fed mice triggers diabetes by disrupting beta cell capacity
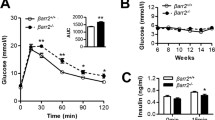
Adult mice with specific Xbp1 deletion in beta cells (Xbp1flox/flox Pdx1-CreER, denoted β-Xbp1−/−) and littermate control mice (Xbp1+/+ Pdx1-CreER, denoted β-Xbp1+/+) were fed a chow or a high-fat diet for 5 weeks. Body weight was not different among the groups (ESM Fig. 2). Morning fed blood glucose levels in chow-fed β-Xbp1−/− mice were not different from chow- and high-fat-fed β-Xbp1+/+ mice throughout the study (Fig. 1a). In contrast, the blood glucose levels in high-fat-fed β-Xbp1−/− mice were significantly increased by 2 weeks after commencement of the diet and remained significantly higher compared with all the other groups throughout the study (Fig. 1a). The AUC for blood glucose levels from 0 to 90 min during OGTT was significantly increased in chow-fed β-Xbp1−/− mice compared with β-Xbp1+/+ mice (Fig. 1b), indicating that the adult-onset deletion of Xbp1 impairs glucose tolerance. After high-fat feeding, β-Xbp1−/− mice displayed an elevated fasting glucose and severely worsened glucose tolerance compared with all other groups (Fig. 1b). These findings demonstrate that the deletion of Xbp1 in beta cells of adult mice that are normally resistant to the development of diabetes induces glucose intolerance, with severe worsening and the development of diabetes when challenged by high-fat diet feeding.

Beta cell-specific Xbp1 deletion in adult mice fed a high-fat diet triggers diabetes by disrupting insulin secretory capacity. (a) Fed blood glucose levels. (b) Blood glucose levels and AUC for glucose during OGTT. (c) Serum insulin levels and AUC for insulin during OGTT. (d) Insulin secretion from isolated islets treated with low (2 mmol/l) or high (20 mmol/l) stimulatory level of glucose. (e) Insulin content in islets. (f) mRNA expression of insulin and Pc2 in islets expressed as fold change of the levels in chow-fed β-Xbp1+/+ mice. (g) Serum proinsulin levels. (h) mRNA expression of adaptive UPR genes in islets expressed as fold change of the levels in chow-fed β-Xbp1+/+ mice. (i) mRNA expression of pro-apoptosis UPR genes in islets expressed as fold change of the levels in chow-fed β-Xbp1+/+ mice. (j) mRNA expression of beta cell function genes in islets expressed as fold change of the levels in chow-fed β-Xbp1+/+ mice. All data are represented as means ± SEM. n = 5–11, chow-fed β-Xbp1+/+; n = 6–19, high-fat-fed β-Xbp1+/+; n = 5–16, chow-fed β-Xbp1−/−; n = 6–18, high-fat-fed β-Xbp1−/−. ANOVA: *p<0.05, **p<0.01, ***p<0.001 genotype effect; †p<0.05, ††p<0.01, †††p<0.001 diet effect. C, chow-fed; HF, high-fat-fed; Tam., tamoxifen
The AUC for insulin levels during the OGTT was markedly reduced in high-fat-fed β-Xbp1−/− mice compared with β-Xbp1+/+ mice (Fig. 1c). This shows that the deterioration of glucose tolerance in β-Xbp1−/− mice after high-fat feeding was associated with lowered plasma insulin levels.
Compared with chow-fed mice, insulin secretion at a high stimulatory level of glucose (20 mmol/l) was increased in islets isolated from high-fat-fed β-Xbp1+/+ mice (Fig. 1d). Additionally, insulin content was increased in islets from high-fat-fed β-Xbp1+/+ mice compared with chow-fed controls (Fig. 1e). These findings are consistent with the usual enhancement of beta cell insulin secretory capacity in response to high-fat diet-induced insulin resistance. There were no differences detected in insulin secretion at both low and high stimulatory levels of glucose between islets from chow-fed β-Xbp1+/+ and β-Xbp1−/− mice (Fig. 1d). However, after high-fat feeding, islets of β-Xbp1−/− mice displayed significantly reduced insulin secretion at both low and high glucose levels compared with β-Xbp1+/+ mice (Fig. 1d). Furthermore, insulin content was reduced in islets from β-Xbp1−/− mice, with no difference between the diets (Fig. 1e). This was associated with significantly reduced insulin mRNA levels in islets of β-Xbp1−/− mice compared with β-Xbp1+/+ mice, with no effects of diet (Fig. 1f). Ultrastructure analysis by transmission electron microscopy (ESM Methods) revealed that some beta cells from β-Xbp1−/− mice displayed fewer insulin granules compared with β-Xbp1+/+ mice, irrespective of diet (ESM Fig. 3). Serum proinsulin levels were increased in β-Xbp1−/− mice fed a chow diet, and were increased further after high-fat feeding (Fig. 1g). The mRNA levels of the proinsulin processing enzyme, prohormone convertase-2 (Pc2, also known as Pcsk2), were increased in islets from β-Xbp1+/+ mice fed a high-fat diet, whereas they were reduced in islets from β-Xbp1−/− mice after high-fat feeding (Fig. 1f). These data suggest that Xbp1 deletion lowers islet insulin content, impairs proinsulin processing and disrupts the capacity of beta cells for insulin secretory adaptation in response to high-fat feeding, ultimately resulting in diabetes.
XBP1 maintains expression of genes involved in the UPR and beta cell function
The expression of Xbp1 and adaptive UPR genes, including ER chaperones, foldases and ER-associated degradation (ERAD) components [Bip (also known as Hspa5), Erp72 (also known as Pdia4), Edem1 and Fkbp11], and Xbp1 splicing were reduced in islets from β-Xbp1−/− mice compared with islets from β-Xbp1+/+ mice, with no differences between the diets (Fig. 1h). The expression of the pro-apoptotic UPR gene, Chop (also known as Ddit3), was not different between genotypes or diets, whereas Trib3 expression was significantly increased in islets of β-Xbp1−/− mice compared with β-Xbp1+/+ mice fed either a chow or high-fat diet (Fig. 1i).
We examined the expression of genes important for beta cell function. The glucose transporter, Glut2 (also known as Slc2a2), was significantly downregulated in the islets of β-Xbp1−/− mice compared with control β-Xbp1+/+ mice fed either a chow or a high-fat diet (Fig. 1j). However, the mRNA levels of the other beta cell function genes assessed [Gck, Pcx, Gpd2, Kir6.2 (also known as Kcnj11), Gpr40 (also known as Ffar1), Glp1r and Gipr] were reduced solely in islets of β-Xbp1−/− mice fed a high-fat diet (Fig. 1j). These findings suggest that XBP1 is required to maintain the expression of genes important for beta cell function in mice challenged by a high-fat diet.
Xbp1 deletion alters islet cell composition and enhances beta cell turnover
Total pancreas weights were not different between β-Xbp1+/+ and β-Xbp1−/− mice, irrespective of diet (Fig. 2a). Beta cell mass was significantly increased in high-fat-fed β-Xbp1+/+ mice compared with chow-fed controls (Fig. 2b). However, relative to high-fat-fed β-Xbp1+/+ mice, beta cell mass was reduced in high-fat-fed β-Xbp1−/− mice, with values similar to those observed in chow-fed β-Xbp1+/+ mice (Fig. 2b). A non-significant increase in beta cell mass was apparent in chow-fed β-Xbp1−/− mice compared with chow-fed controls (Fig. 2b). Interestingly, the rate of beta cell proliferation, assessed using Ki-67 immunostaining, was significantly increased in β-Xbp1−/− mice compared with β-Xbp1+/+ mice, irrespective of diet (Fig. 2c). The rate of beta cell apoptosis, assessed using TUNEL staining, was unchanged in chow-fed β-Xbp1−/− mice, but was significantly increased in β-Xbp1−/− mice after high-fat feeding (Fig. 2d). These findings suggest that Xbp1 deletion leads to beta cell proliferation and expansion under control conditions, but to high beta cell turnover after high-fat feeding, with the increase in beta cell proliferation outstripped by apoptosis, resulting in a relative decline of beta cell mass.

Xbp1 deletion leads to altered islet cell composition, increased beta cell turnover, beta cell dedifferentiation and beta-to-alpha cell transdifferentiation. (a) Pancreas weight. (b) Beta cell mass (quantification of immunostaining for insulin). (c) Beta cell proliferation rate (quantification of immunostaining for Ki-67 and insulin). (d) Beta cell apoptosis rate (quantification of immunostaining for TUNEL and insulin). (e) Representative images of immunostaining for insulin and glucagon. (f) Alpha cell mass (quantification of immunostaining for glucagon). (g) mRNA expression of genes involved in beta cell proliferation (Myc), dedifferentiation (Sox9) and senescence (p21 and p53) in islets, expressed as fold change of the levels in chow-fed β-Xbp1+/+ mice. (h) Representative images of immunostaining for GFP (green), glucagon (red) and insulin (magenta). Scale bar, 20 μm. (i) Expression in immunoprecipitated mRNA of beta cell identity (Pdx1, Beta2, Nkx6.1 and Foxo1) genes expressed as fold change of the levels in β-Xbp1+/+Gt mice, and beta cell dedifferentiation (Aldh1a3) and alpha cell (Arx, Irx2 and Gcg) genes expressed as fold change of the levels in β-Xbp1−/−Gt mice. All data are represented as means ± SEM. (a–g) n = 4–8, chow-fed β-Xbp1+/+; n = 6–10, high-fat-fed β-Xbp1+/+; n = 4–7, chow-fed β-Xbp1−/−; n = 6–9, high-fat-fed β-Xbp1−/−. ANOVA: *p<0.05, **p<0.01, ***p<0.001 genotype effect; †p<0.05, ††p<0.01 diet effect. (h, i) n = 3–7, β-Xbp1+/+Gt; n = 3–8, β-Xbp1−/−Gt. Unpaired two-tailed t test: *p<0.05, **p<0.01, ***p<0.001. C, chow-fed; HF, high-fat-fed. Gluc, glucagon; Ins, insulin
Whereas the β-Xbp1+/+ mice showed a typical organisation of mouse islets with a beta cell core and alpha cells distributed around the mantle, the islet pattern in β-Xbp1−/− mice was often altered, with alpha cells distributed throughout the islets (Fig. 2e). Strikingly, alpha cell mass was increased by ~2-fold in β-Xbp1−/− mice compared with β-Xbp1+/+ mice fed a chow diet, but was not different between the genotypes after high-fat feeding (Fig. 2f).
Xbp1 deletion increases expression of cell proliferation, senescence and inflammation markers
The expression of Myc, a cell cycle regulator [32], was increased in islets from β-Xbp1−/− mice fed either a chow or a high-fat diet (Fig. 2g) in parallel with enhanced beta cell proliferation. Furthermore, the expression levels of the pancreatic progenitor and beta cell dedifferentiation marker, Sox9 [33], cell cycle/senescence markers, p21 (also known as Cdkn1a) and p53 (also known as Trp53) (Fig. 2g), inflammatory cytokines (Il6, Il1b and Tnf) and the macrophage marker, F4/80 (also known as Adgre1) (ESM Fig. 4), were significantly increased in islets from high-fat-fed β-Xbp1−/− mice. Reduced insulin production can induce beta cell proliferation [14].
Xbp1 deletion leads to beta cell dedifferentiation and beta-to-alpha cell transdifferentiation
We next deleted Xbp1 in adult beta cells and tracked their fate using a gene trap Rosa26GFP lineage label in Xbp1flox/flox Pdx1-CreER Gt/Rosa26GFP (β-Xbp1−/−Gt) and control Xbp1+/+ Pdx1-CreER Gt/Rosa26GFP (β-Xbp1+/+Gt) mice (ESM Fig. 5). Five weeks after tamoxifen administration, GFP staining was co-localised with insulin and not with glucagon in pancreases of β-Xbp1+/+Gt mice, as expected (Fig. 2h). However, in β-Xbp1−/−Gt mice, some cells stained for GFP but not for insulin, and substantial co-staining of GFP and glucagon was observed (Fig. 2h). This shows that deletion of Xbp1 in adult beta cells leads to both a loss of insulin and the production of glucagon. The conversion of beta cells to glucagon-producing cells provides a mechanism for increased alpha cell mass in mice with Xbp1 deletion.
We then used gene trap mice to conduct TRAP experiments. GFP+ ribosomal complexes were purified from islets isolated from chow-fed β-Xbp1+/+Gt and β-Xbp1−/−Gt mice in which the GFP–L10a ribosomal fusion protein is produced only in Pdx1-CreER-labelled beta cells and derived cells. Using quantitative PCR analysis, Xbp1s mRNA levels were shown to be significantly reduced in islets from β-Xbp1−/−Gt mice compared with control β-Xbp1+/+Gt mice (Fig. 2i). We then examined the expression of four transcription factors that are critical for the maintenance of beta cell identity in adult islets. Notably, Pdx1, Beta2 (also known as Neurod1), Nkx6.1 and Foxo1 were significantly reduced in the islets of β-Xbp1−/−Gt mice compared with control mice (Fig. 2i). This was associated with an 82% reduction in insulin mRNA levels in β-Xbp1−/−Gt mice, whereas expression of the beta cell dedifferentiation marker Aldh1a3 [8] was markedly increased (Fig. 2i). Next, we assessed the expression of genes restricted to alpha cells in adult animals. Strikingly, Arx, Irx2 and glucagon (Gcg) mRNA levels were markedly increased in β-Xbp1−/−Gt mice compared with control mice (Fig. 2i). This suggests that Xbp1 deletion in adult beta cells leads to beta-to-alpha cell reprogramming.
Xbp1 deletion in beta cells of obese ob/ob mice leads to failure of beta cell compensation and diabetes
To test whether XBP1 expression makes a necessary contribution to in vivo beta cell compensation and diabetes resistance in obese mice, we crossed Xbp1flox/flox Pdx1-CreER and ob/+ mice to generate Xbp1flox/flox Pdx1-CreER ob/ob (β-Xbp1−/−Ob) and Xbp1flox/flox Pdx1-CreER (β-Xbp1−/−Wt) mice and control Xbp1+/+ Pdx1-CreER ob/ob (β-Xbp1+/+Ob) and Xbp1+/+ Pdx1-CreER (β-Xbp1+/+Wt) mice. Xbp1 deletion was induced in 8–10-week-old mice by tamoxifen administration. Body weight was increased in the groups of ob/ob mice (β-Xbp1+/+Ob and β-Xbp1−/−Ob) compared with respective lean controls (β-Xbp1+/+Wt and β-Xbp1−/−Wt), but was not affected by Xbp1 deletion (ESM Fig. 6). Fed blood glucose levels in obese β-Xbp1−/−Ob mice were significantly increased 1 and 2 weeks after Xbp1 deletion compared with all other groups (Fig. 3a), demonstrating that Xbp1 deletion leads to diabetes under conditions of obesity. Obese β-Xbp1+/+Ob mice compared with lean β-Xbp1+/+Wt mice, as expected, were glucose intolerant, as shown by the OGTT blood glucose levels and glucose AUC (Fig. 3b). Elevated fasting glucose and more severe glucose intolerance were observed in β-Xbp1−/−Ob mice (Fig. 3b). Insulin levels and the insulin AUC during the OGTT were significantly increased in obese β-Xbp1+/+Ob mice compared with lean β-Xbp1+/+Wt mice (Fig. 3c), characteristic of the beta cell compensatory response to obesity. Insulin excursions, however, were significantly reduced in β-Xbp1−/−Ob mice compared with β-Xbp1+/+Ob mice (Fig. 3c). Xbp1 deletion may therefore result in impaired compensatory insulin secretion under conditions of obesity, causing diabetes.

Beta cell-specific Xbp1 deletion in obese ob/ob mice leads to diabetes due to failure of beta cell compensation. (a) Fed blood glucose levels. (b) Blood glucose levels and AUC for glucose during OGTT. (c) Serum insulin levels and AUC for insulin during OGTT. (d) mRNA expression of adaptive UPR genes in islets expressed as fold change of the levels in β-Xbp1+/+Wt mice. (e) mRNA expression of pro-apoptosis UPR genes in islets expressed as fold change of the levels in β-Xbp1+/+Wt mice. (f) mRNA expression of insulin and Pc2 in islets expressed as fold change of the levels in β-Xbp1+/+Wt mice. (g) Serum proinsulin levels. (h) Insulin secretion from isolated islets treated with low (2 mmol/l) or high (20 mmol/l) stimulatory level of glucose for 1 h. (i) Insulin content in islets. (j) Insulin secretion from isolated islets of β-Xbp1+/+Ob and β-Xbp1−/−Ob mice treated with repeated low (2 mmol/l) or high (20 mmol/l) stimulatory level of glucose for three cycles. (k) Insulin content in islets following three cycles of glucose treatment. (l) Representative images of TUNEL immunostaining in pancreas sections. (m) Beta cell apoptosis rate (quantification of immunostaining for TUNEL and insulin). All data are represented as means ± SEM. (a–i) n = 3–12, β-Xbp1+/+Wt; n = 3–14, β-Xbp1−/−Wt; n = 3–7, β-Xbp1+/+Ob; n = 7–13, β-Xbp1−/−Ob. (j, k) n = 5, β-Xbp1+/+Ob; n = 4, β-Xbp1−/−Ob. (l, m) n = 6, β-Xbp1+/+Wt; n = 6, β-Xbp1−/−Wt; n = 6, β-Xbp1+/+Ob; n = 8, β-Xbp1−/−Ob. ANOVA: *p<0.05, **p<0.01, ***p<0.001 Xbp1 genotype effect; †p<0.05, ††p<0.01, †††p<0.001 Ob genotype effect. Tam., tamoxifen
In islets isolated from obese β-Xbp1+/+Ob mice, the mRNA levels of Xbp1 and the adaptive UPR genes were increased compared with lean β-Xbp1+/+Wt mice (Fig. 3d), thus confirming that adaptive UPR activation is associated with beta cell compensation in obese mice. As expected, the expression levels of Xbp1 and spliced Xbp1 were significantly reduced in islets of β-Xbp1−/−Wt and β-Xbp1−/−Ob mice compared with respective control mice (Fig. 3d). This was associated with downregulation of Bip, Erp72, Edem1 and Fkbp11 (Fig. 3d), thus demonstrating that Xbp1 deletion reduces expression of adaptive UPR genes in islets of lean mice and results in their impaired activation in islets of obese mice. On the other hand, the expression levels of the deleterious UPR genes, Chop and Trib3, were increased in islets of β-Xbp1−/−Wt compared with β-Xbp1+/+Wt mice, with suggestion of partial attenuation of this effect in β-Xbp1−/−Ob mice (Fig. 3e). Thus, unexpectedly, the inhibition of XBP1-mediated adaptive UPR in islets of obese β-Xbp1−/−Ob mice was not associated with a pro-apoptotic UPR activation (Fig. 3e).
The expression of insulin and Pc2 was significantly induced in the islets of obese β-Xbp1+/+Ob mice compared with lean β-Xbp1+/+Wt mice (Fig. 3f), providing evidence of an improved beta cell capacity to meet greater insulin demand in obesity. However, insulin and Pc2 mRNA levels were significantly reduced following Xbp1 deletion in both lean β-Xbp1−/−Wt and obese β-Xbp1−/−Ob mice (Fig. 3f). The highest levels of serum proinsulin among the groups were observed in obese β-Xbp1−/−Ob mice (Fig. 3g), thus confirming the significance of diminished beta cell capacity for proinsulin processing in the setting of high insulin demand.
The expected compensatory increase in glucose-stimulated insulin secretion in obesity was observed in islets isolated from β-Xbp1+/+Ob mice (Fig. 3h). Surprisingly, this response was enhanced in islets from β-Xbp1−/−Ob mice (Fig. 3h), implying that under static ex vivo conditions secretory mechanisms may be preserved with Xbp1 deletion. We hypothesised that challenge with a single glucose exposure might not be sufficient to reveal limitations on secretion. We therefore subjected islets from β-Xbp1+/+Ob and β-Xbp1−/−Ob mice to repeated high-glucose exposure to mimic the metabolic challenge caused by hyperphagia in obese mice [34]. After three cycles of high-glucose stimulation, β-Xbp1+/+Ob islets displayed only a slight non-significant decline in insulin secretion, whereas insulin secretion in β-Xbp1−/−Ob islets was significantly reduced by ~50% (Fig. 3j), with unchanged insulin content (Fig. 3k). Similar results were obtained using β-Xbp1−/−Wt islets, although with reduced insulin content (ESM Fig. 7). Thus, islets with Xbp1 deletion were susceptible to insulin secretory dysfunction after repeated metabolic challenge.
Xbp1 deletion in beta cells of ob/ob mice leads to increased beta cell apoptosis
The incidence of TUNEL-positive beta islets in obese β-Xbp1+/+Ob mice was not different from control β-Xbp1+/+Wt mice, suggesting that beta cell apoptosis was not altered in diabetes-resistant ob/ob mice (Fig. 3l,m). In contrast, the incidence of TUNEL-positive cells, although unchanged in β-Xbp1−/−Wt mice, was significantly increased by threefold in β-Xbp1−/−Ob mice (Fig. 3l,m), suggesting that the absence of XBP1 increases the rate of beta cell apoptosis under conditions of obesity. Notably, the findings reveal that the variations in beta cell apoptosis did not correlate with the changes in pro-apoptotic UPR, which raises the important question: what is the mechanism(s) of increased beta cell apoptosis following XBP1 loss under conditions of metabolic challenge?
Xbp1 deletion increases beta cell apoptosis by attenuating the antioxidant response under metabolic stress conditions
To address this question, we established a model of ex vivo glucolipotoxicity using β-Xbp1+/+ and β-Xbp1−/− islets treated with high glucose (25 mmol/l) + saturated fatty acid palmitate (0.4 mmol/l coupled to 0.92% BSA) for 72 h (termed GP). Notably, the twofold increase in apoptosis observed in β-Xbp1+/+ islets after GP exposure was significantly potentiated in β-Xbp1−/− islets (Fig. 4a). The expression levels of spliced Xbp1 and the adaptive UPR genes, Bip, Erp72, Edem1 and Fkbp11, were increased after GP exposure of β-Xbp1+/+ islets (Fig. 4b). However, in β-Xbp1−/− islets, the expression levels of Xbp1 and spliced Xbp1 were significantly reduced along with the adaptive UPR genes in islets treated under control conditions, and their activation by GP treatment was markedly attenuated (Fig. 4b). The mRNA levels of the deleterious UPR genes, Chop, Atf3 and Trib3, were strongly increased by GP treatment in control β-Xbp1+/+ islets, whereas a comparatively blunted response was observed in β-Xbp1−/− islets (Fig. 4c). These results with ex vivo glucolipoapoptosis closely resemble the in vivo findings, suggesting that the potentiation of beta cell apoptosis following XBP1 loss in metabolic stress conditions occurs in concert with lowered pro-apoptotic UPR.

Xbp1 deletion in beta cells potentiates glucolipoapoptosis by attenuating the antioxidant response. (a) Apoptosis rate in islets determined by cell death detection ELISA. (b) mRNA expression of adaptive UPR genes in islets expressed as fold change of the levels in β-Xbp1+/+ islets. (c) mRNA expression of pro-apoptosis UPR genes in islets expressed as fold change of the levels in β-Xbp1+/+ islets. (d) mRNA expression of antioxidant enzymes in islets expressed as fold change of the levels in β-Xbp1+/+ islets. (e) Apoptosis rate in islets co-treated in the absence or presence of the antioxidant. Islets were treated in the absence (control, C) or presence of high GP for 72 h. All data are represented as means±SEM. n = 6–9, β-Xbp1+/+; n = 8–9, β-Xbp1−/−. ANOVA: *p<0.05, **p<0.01, ***p<0.001 genotype effect; †p<0.05, ††p<0.01, †††p<0.001 treatment effect
Oxidative stress has been implicated in beta cell apoptosis in type 2 diabetes [35] and has been linked with UPR deregulation [36]. We therefore examined whether changes in redox status could contribute to the potentiation of glucolipoapoptosis in β-Xbp1−/− islets. GP treatment significantly increased the expression of several antioxidant enzymes, heme-oxygenase-1 (Hmox1), superoxide dismutase-1 (Sod1) and glutathione peroxidase (Gpx1), in β-Xbp1+/+ islets (Fig. 4d), consistent with activation of a robust adaptive response to oxidative stress. In contrast, the antioxidant mRNA levels were significantly reduced in GP-treated β-Xbp1−/− islets compared with β-Xbp1+/+ islets (Fig. 4d), suggesting that Xbp1 deletion inhibited the antioxidant response. We next co-treated islets with NAC, a commonly used antioxidant that acts as a reduced glutathione precursor [37]. NAC co-treatment completely rescued β-Xbp1−/− islets from the potentiation of glucolipoapoptosis (Fig. 4e). These findings suggest that XBP1 protects against beta cell apoptosis during metabolic stress by promoting an antioxidant response.
Discussion
Our data demonstrate that XBP1 plays an essential role in the maintenance of beta cell identity, in the repression of beta-to-alpha cell reprogramming and in preserving normoglycaemia in the face of insulin resistance by facilitating beta cell adaptation. Loss of XBP1 resulted in beta cell dedifferentiation, as evidenced by: (1) downregulation of beta cell-enriched genes; (2) concomitant upregulation of beta cell forbidden genes; and (3) appearance of alpha cell features in lineage-labelled beta cells. The maladaptation of XBP1-deficient beta cells under metabolic stress conditions was associated with deterioration of beta cell dysfunction and increased beta cell apoptosis due to impaired handling of oxidative stress. Thus, our studies suggest that the decline in spliced XBP1 expression found in type 2 diabetes [29] contributes to the failure of beta cells to secrete sufficient amounts of insulin to meet metabolic demand.
Primary defects induced by XBP1 deficiency in the absence of diabetes and, thus, independently of glucotoxicity, include beta cell dedifferentiation, beta-to-alpha cell conversion and impaired proinsulin processing. These underlying defects rendered beta cells incapable of adapting to metabolic challenge, triggering hyperglycaemia and further decline of beta cell capacity. Glucotoxicity [38], inflammatory stress [39] and oxidative stress [35] may play important roles in beta cell deterioration and development of secondary abnormalities, which include islet inflammation, cellular senescence and increased beta cell apoptosis.
Previous studies showed that embryonic deletion of Xbp1 in beta cells led to reduced islet area in association with impaired beta cell proliferation [21]. In stark contrast, the deletion of Xbp1 in adult beta cells resulted in increased proliferation, while beta cell loss under metabolic stress conditions was due to increased apoptosis. Moreover, previous studies in MIN6 cells led to the conclusion that XBP1 deficiency resulted in feedback inositol-requiring enzyme 1 α (IRE1α) hyperactivation [21]. However, we found unchanged IRE1α phosphorylation in islets isolated from Xbp1 knockout mice fed a chow diet compared with control mice (ESM Fig. 8). Furthermore, the expression of Atf4, a transcription factor induced downstream of PERK, was unchanged (ESM Fig. 9). Instead, our data demonstrate that beta cell dedifferentiation induced by Xbp1 deletion was associated with altered expression of multiple transcriptional regulators of beta cell identity (Pdx1, Beta2, Nkx6.1 and Foxo1) [8, 40,41,42,43,44,45,46], and increased expression of Myc, a cell cycle regulator that promotes beta cell replication, but concomitantly diverts beta cells towards an immature phenotype [32, 47, 48]. Chronic hyperglycaemia, islet cell aggregation and several specific genetic manipulations can lead to conversion of beta cells to alpha cells [8, 10, 11, 46, 49,50,51,52,53,54,55,56,57]. To our knowledge, this is the first study showing that a UPR factor can drive the process of beta-to-alpha cell transdifferentiation.
IRE1α has been implicated in the regulation of insulin biosynthesis [27, 58,59,60]. Compared with our model of Xbp1 deletion, IRE1α deletion in beta cells of adult mice caused a significantly greater diabetic phenotype in association with increased production of reactive oxygen species, but without reducing expression of beta cell-specific mRNAs [60]. Moreover, a recent study showed that IRE1α deletion in the non-obese diabetic (NOD) mouse model of type 1 diabetes induced transient hyperglycaemia and beta cell dedifferentiation, which protected against immune-mediated destruction [61]. Our study highlights a requirement of XBP1 in activation of antioxidant genes that promote beta cell survival under metabolic stress conditions.
In summary, we present the first evidence that XBP1 maintains mature beta cell identity, represses beta-to-alpha cell transdifferentiation and is required for beta cell protection against diabetes in insulin resistance states. From a therapeutic perspective, targeting XBP1 might help to reverse the process of beta cell dedifferentiation and restore functional beta cell mass in type 2 diabetes.
Data availability
All data generated and analysed during this study are included in this published article (and its supplementary material files).
Abbreviations
- ER:
-
Endoplasmic reticulum
- GP:
-
Glucose + palmitate
- IRE1:
-
Inositol-requiring enzyme 1
- IRE1α:
-
Inositol-requiring enzyme 1 α
- NAC:
-
N-acetyl-l-cysteine
- PERK:
-
PKR-like ER kinase
- TRAP:
-
Translating ribosome affinity purification
- UPR:
-
Unfolded protein response
- XBP1:
-
X-box binding protein 1
References
Chen C, Cohrs CM, Stertmann J, Bozsak R, Speier S (2017) Human beta cell mass and function in diabetes: recent advances in knowledge and technologies to understand disease pathogenesis. Mol Metab 6(9):943–957. https://doi.org/10.1016/j.molmet.2017.06.019
Esser N, Utzschneider KM, Kahn SE (2020) Early beta cell dysfunction vs insulin hypersecretion as the primary event in the pathogenesis of dysglycaemia. Diabetologia 63(10):2007–2021. https://doi.org/10.1007/s00125-020-05245-x
Prentki M, Nolan CJ (2006) Islet beta cell failure in type 2 diabetes. J Clin Invest 116(7):1802–1812. https://doi.org/10.1172/JCI29103
Cohrs CM, Panzer JK, Drotar DM et al (2020) Dysfunction of persisting beta cells is a key feature of early type 2 diabetes pathogenesis. Cell Rep 31(1):107469. https://doi.org/10.1016/j.celrep.2020.03.033
Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC (2003) Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52(1):102–110. https://doi.org/10.2337/diabetes.52.1.102
Bensellam M, Jonas JC, Laybutt DR (2018) Mechanisms of beta-cell dedifferentiation in diabetes: recent findings and future research directions. J Endocrinol 236(2):R109–R143. https://doi.org/10.1530/JOE-17-0516
Jonas JC, Sharma A, Hasenkamp W et al (1999) Chronic hyperglycemia triggers loss of pancreatic beta cell differentiation in an animal model of diabetes. J Biol Chem 274:14112–14121. https://doi.org/10.1074/jbc.274.20.14112
Talchai C, Xuan S, Lin HV, Sussel L, Accili D (2012) Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 150(6):1223–1234. https://doi.org/10.1016/j.cell.2012.07.029
Sun J, Ni Q, Xie J et al (2019) beta-cell dedifferentiation in patients with T2D with adequate glucose control and nondiabetic chronic pancreatitis. J Clin Endocrinol Metab 104(1):83–94. https://doi.org/10.1210/jc.2018-00968
Brereton MF, Iberl M, Shimomura K et al (2014) Reversible changes in pancreatic islet structure and function produced by elevated blood glucose. Nat Commun 5:4639. https://doi.org/10.1038/ncomms5639
Wang Z, York NW, Nichols CG, Remedi MS (2014) Pancreatic beta cell dedifferentiation in diabetes and redifferentiation following insulin therapy. Cell Metab 19(5):872–882. https://doi.org/10.1016/j.cmet.2014.03.010
Kjorholt C, Akerfeldt MC, Biden TJ, Laybutt DR (2005) Chronic hyperglycemia, independent of plasma lipid levels, is sufficient for the loss of beta-cell differentiation and secretory function in the db/db mouse model of diabetes. Diabetes 54(9):2755–2763. https://doi.org/10.2337/diabetes.54.9.2755
Liu M, Weiss MA, Arunagiri A et al (2018) Biosynthesis, structure, and folding of the insulin precursor protein. Diabetes Obes Metab 20(Suppl 2):28–50. https://doi.org/10.1111/dom.13378
Szabat M, Page MM, Panzhinskiy E et al (2016) Reduced insulin production relieves endoplasmic reticulum stress and induces beta cell proliferation. Cell Metab 23(1):179–193. https://doi.org/10.1016/j.cmet.2015.10.016
Sharma RB, O'Donnell AC, Stamateris RE et al (2015) Insulin demand regulates beta cell number via the unfolded protein response. J Clin Invest 125(10):3831–3846. https://doi.org/10.1172/JCI79264
Sharma RB, Snyder JT, Alonso LC (2019) Atf6alpha impacts cell number by influencing survival, death and proliferation. Mol Metab 27(Suppl):S69–S80. https://doi.org/10.1016/j.molmet.2019.06.005
Chan JY, Luzuriaga J, Bensellam M, Biden TJ, Laybutt DR (2013) Failure of the adaptive unfolded protein response in islets of obese mice is linked with abnormalities in beta-cell gene expression and progression to diabetes. Diabetes 62(5):1557–1568. https://doi.org/10.2337/db12-0701
Chan JY, Luzuriaga J, Maxwell EL, West PK, Bensellam M, Laybutt DR (2015) The balance between adaptive and apoptotic unfolded protein responses regulates beta-cell death under ER stress conditions through XBP1, CHOP and JNK. Mol Cell Endocrinol 413:189–201. https://doi.org/10.1016/j.mce.2015.06.025
Eizirik DL, Pasquali L, Cnop M (2020) Pancreatic beta-cells in type 1 and type 2 diabetes mellitus: different pathways to failure. Nat Rev Endocrinol 16(7):349–362. https://doi.org/10.1038/s41574-020-0355-7
Herbert TP, Laybutt DR (2016) A reevaluation of the role of the unfolded protein response in islet dysfunction: maladaptation or a failure to adapt? Diabetes 65(6):1472–1480. https://doi.org/10.2337/db15-1633
Lee AH, Heidtman K, Hotamisligil GS, Glimcher LH (2011) Dual and opposing roles of the unfolded protein response regulated by IRE1alpha and XBP1 in proinsulin processing and insulin secretion. Proc Natl Acad Sci U S A 108(21):8885–8890. https://doi.org/10.1073/pnas.1105564108
Cunha DA, Gurzov EN, Naamane N et al (2014) JunB protects beta-cells from lipotoxicity via the XBP1-AKT pathway. Cell Death Differ 21(8):1313–1324. https://doi.org/10.1038/cdd.2014.53
Allagnat F, Christulia F, Ortis F et al (2010) Sustained production of spliced X-box binding protein 1 (XBP1) induces pancreatic beta cell dysfunction and apoptosis. Diabetologia 53(6):1120–1130. https://doi.org/10.1007/s00125-010-1699-7
Moore BD, Jin RU, Lo H et al (2016) Transcriptional regulation of X-box-binding protein one (XBP1) by hepatocyte nuclear factor 4alpha (HNF4Alpha) is vital to Beta-cell function. J Biol Chem 291(12):6146–6157. https://doi.org/10.1074/jbc.M115.685750
Kirkpatrick CL, Wiederkehr A, Baquie M et al (2011) Hepatic nuclear factor 1alpha (HNF1alpha) dysfunction down-regulates X-box-binding protein 1 (XBP1) and sensitizes beta-cells to endoplasmic reticulum stress. J Biol Chem 286(37):32300–32312. https://doi.org/10.1074/jbc.M111.247866
Sharma RB, Darko C, Alonso LC (2020) Intersection of the ATF6 and XBP1 ER stress pathways in mouse islet cells. J Biol Chem 295(41):14164–14177. https://doi.org/10.1074/jbc.RA120.014173
Tsuchiya Y, Saito M, Kadokura H et al (2018) IRE1-XBP1 pathway regulates oxidative proinsulin folding in pancreatic beta cells. J Cell Biol 217(4):1287–1301. https://doi.org/10.1083/jcb.201707143
Takatani T, Shirakawa J, Roe MW et al (2016) IRS1 deficiency protects beta-cells against ER stress-induced apoptosis by modulating sXBP-1 stability and protein translation. Sci Rep 6:28177. https://doi.org/10.1038/srep28177
Engin F, Nguyen T, Yermalovich A, Hotamisligil GS (2014) Aberrant islet unfolded protein response in type 2 diabetes. Sci Rep 4:4054. https://doi.org/10.1038/srep04054
Heiman M, Kulicke R, Fenster RJ, Greengard P, Heintz N (2014) Cell type-specific mRNA purification by translating ribosome affinity purification (TRAP). Nat Protoc 9(6):1282–1291. https://doi.org/10.1038/nprot.2014.085
Ip CK, Zhang L, Farzi A et al (2019) Amygdala NPY circuits promote the development of accelerated obesity under chronic stress conditions. Cell Metab 30(1):111–128.e6. https://doi.org/10.1016/j.cmet.2019.04.001
Puri S, Roy N, Russ HA et al (2018) Replication confers beta cell immaturity. Nat Commun 9(1):485. https://doi.org/10.1038/s41467-018-02939-0
Puri S, Akiyama H, Hebrok M (2013) VHL-mediated disruption of Sox9 activity compromises beta-cell identity and results in diabetes mellitus. Genes Dev 27(23):2563–2575. https://doi.org/10.1101/gad.227785.113
Kebede MA, Oler AT, Gregg T et al (2014) SORCS1 is necessary for normal insulin secretory granule biogenesis in metabolically stressed beta cells. J Clin Invest 124(10):4240–4256. https://doi.org/10.1172/JCI74072
Roma LP, Jonas JC (2020) Nutrient metabolism, subcellular redox state, and oxidative stress in pancreatic islets and beta-cells. J Mol Biol 432(5):1461–1493. https://doi.org/10.1016/j.jmb.2019.10.012
Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ (2008) Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J Clin Invest 118(10):3378–3389. https://doi.org/10.1172/JCI34587
Tanaka Y, Tran PO, Harmon J, Robertson RP (2002) A role for glutathione peroxidase in protecting pancreatic beta cells against oxidative stress in a model of glucose toxicity. Proc Natl Acad Sci U S A 99(19):12363–12368. https://doi.org/10.1073/pnas.192445199
Weir GC (2020) Glucolipotoxicity, beta-cells, and diabetes: the emperor has no clothes. Diabetes 69(3):273–278. https://doi.org/10.2337/db19-0138
Donath MY, Dinarello CA, Mandrup-Poulsen T (2019) Targeting innate immune mediators in type 1 and type 2 diabetes. Nat Rev Immunol 19(12):734–746. https://doi.org/10.1038/s41577-019-0213-9
Waeber G, Thompson N, Nicod P, Bonny C (1996) Transcriptional activation of the GLUT2 gene by the IPF-1/STF-1/IDX-1 homeobox factor. Mol Endocrinol 10:1327–1334
Ahlgren U, Jonsson J, Jonsson L, Simu K, Edlund H (1998) beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes Dev 12:1763–1768. https://doi.org/10.1101/gad.12.12.1763
Kim JW, Seghers V, Cho JH et al (2002) Transactivation of the mouse sulfonylurea receptor I gene by BETA2/NeuroD. Mol Endocrinol 16(5):1097–1107. https://doi.org/10.1210/mend.16.5.0934
van der Meulen T, Huising MO (2015) Role of transcription factors in the transdifferentiation of pancreatic islet cells. J Mol Endocrinol 54(2):R103–R117. https://doi.org/10.1530/JME-14-0290
Schaffer AE, Taylor BL, Benthuysen JR et al (2013) Nkx6.1 controls a gene regulatory network required for establishing and maintaining pancreatic Beta cell identity. PLoS Genet 9(1):e1003274. https://doi.org/10.1371/journal.pgen.1003274
Taylor BL, Liu FF, Sander M (2013) Nkx6.1 is essential for maintaining the functional state of pancreatic beta cells. Cell Rep 4(6):1262–1275. https://doi.org/10.1016/j.celrep.2013.08.010
Gao T, McKenna B, Li C et al (2014) Pdx1 maintains beta cell identity and function by repressing an alpha cell program. Cell Metab 19(2):259–271. https://doi.org/10.1016/j.cmet.2013.12.002
Robson SC, Ward L, Brown H et al (2011) Deciphering c-MYC-regulated genes in two distinct tissues. BMC Genomics 12:476. https://doi.org/10.1186/1471-2164-12-476
Laybutt DR, Weir GC, Kaneto H et al (2002) Overexpression of c-Myc in beta-cells of transgenic mice causes proliferation and apoptosis, downregulation of insulin gene expression, and diabetes. Diabetes 51:1793–1804. https://doi.org/10.2337/diabetes.51.6.1793
Cinti F, Bouchi R, Kim-Muller JY et al (2016) Evidence of beta-cell dedifferentiation in human type 2 diabetes. J Clin Endocrinol Metab 101(3):1044–1054. https://doi.org/10.1210/jc.2015-2860
Collombat P, Hecksher-Sorensen J, Krull J et al (2007) Embryonic endocrine pancreas and mature beta cells acquire alpha and PP cell phenotypes upon Arx misexpression. J Clin Invest 117(4):961–970. https://doi.org/10.1172/JCI29115
Dhawan S, Georgia S, Tschen SI, Fan G, Bhushan A (2011) Pancreatic beta cell identity is maintained by DNA methylation-mediated repression of Arx. Dev Cell 20(4):419–429. https://doi.org/10.1016/j.devcel.2011.03.012
Papizan JB, Singer RA, Tschen SI et al (2011) Nkx2.2 repressor complex regulates islet beta-cell specification and prevents beta-to-alpha-cell reprogramming. Genes Dev 25(21):2291–2305. https://doi.org/10.1101/gad.173039.111
Gutierrez GD, Bender AS, Cirulli V et al (2017) Pancreatic beta cell identity requires continual repression of non-beta cell programs. J Clin Invest 127(1):244–259. https://doi.org/10.1172/JCI88017
Swisa A, Avrahami D, Eden N et al (2017) PAX6 maintains beta cell identity by repressing genes of alternative islet cell types. J Clin Invest 127(1):230–243. https://doi.org/10.1172/JCI88015
Nishimura W, Takahashi S, Yasuda K (2015) MafA is critical for maintenance of the mature beta cell phenotype in mice. Diabetologia 58(3):566–574. https://doi.org/10.1007/s00125-014-3464-9
Yin Q, Ni Q, Wang Y et al (2020) Raptor determines beta-cell identity and plasticity independent of hyperglycemia in mice. Nat Commun 11(1):2538. https://doi.org/10.1038/s41467-020-15935-0
Spijker HS, Ravelli RB, Mommaas-Kienhuis AM et al (2013) Conversion of mature human beta-cells into glucagon-producing alpha-cells. Diabetes 62(7):2471–2480. https://doi.org/10.2337/db12-1001
Lipson KL, Fonseca SG, Ishigaki S et al (2006) Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metab 4(3):245–254. https://doi.org/10.1016/j.cmet.2006.07.007
Han D, Lerner AG, Vande Walle L et al (2009) IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 138(3):562–575. https://doi.org/10.1016/j.cell.2009.07.017
Hassler JR, Scheuner DL, Wang S et al (2015) The IRE1alpha/XBP1s pathway is essential for the glucose response and protection of beta cells. PLoS Biol 13(10):e1002277. https://doi.org/10.1371/journal.pbio.1002277
Lee H, Lee YS, Harenda Q et al (2020) Beta cell dedifferentiation induced by IRE1alpha deletion prevents type 1 diabetes. Cell Metab 31(4):822–836.e5. https://doi.org/10.1016/j.cmet.2020.03.002
Acknowledgements
We thank the staff of the Garvan Institute Biological Testing Facility and the staff of Australian BioResources for taking care of our test mice. We thank the staff of Garvan Molecular Genetics for genotyping the mice. We also thank A.-H. Lee for reviewing the manuscript.
Author’s relationships and activities
DRL is a member of the Diabetologia Editorial Board. CJN is a member of the Diabetologia Advisory Board. The other authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Funding
Open access funding enabled and organised by CAUL and its Member Institutions. This research was supported by the National Health and Medical Research Council (NHMRC) with project grant 1144206 to DRL.
Author information
Authors and Affiliations
Contributions
KL and DRL conceived and designed experiments, acquired and analysed data and wrote the manuscript. JYC, CL, CKI, Y-CS, HH, WEH, MB, VD-A, MEK and CJN acquired and analysed data and critically reviewed the manuscript. All authors approved the final version of the manuscript. DRL is the guarantor of this work.
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM
(PDF 1077 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, K., Chan, J.Y., Liang, C. et al. XBP1 maintains beta cell identity, represses beta-to-alpha cell transdifferentiation and protects against diabetic beta cell failure during metabolic stress in mice. Diabetologia 65, 984–996 (2022). https://doi.org/10.1007/s00125-022-05669-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-022-05669-7