
Abstract
Aims/hypothesis
Type 2 diabetes is a complex metabolic disease with increasing prevalence worldwide. Improving the prediction of incident type 2 diabetes using epigenetic markers could help tailor prevention efforts to those at the highest risk. The aim of this study was to identify predictive methylation markers for incident type 2 diabetes by combining epigenome-wide association study (EWAS) results from five prospective European cohorts.
Methods
We conducted a meta-analysis of EWASs in blood collected 7–10 years prior to type 2 diabetes diagnosis. DNA methylation was measured with Illumina Infinium Methylation arrays. A total of 1250 cases and 1950 controls from five longitudinal cohorts were included: Doetinchem, ESTHER, KORA1, KORA2 and EPIC-Norfolk. Associations between DNA methylation and incident type 2 diabetes were examined using robust linear regression with adjustment for potential confounders. Inverse-variance fixed-effects meta-analysis of cohort-level individual CpG EWAS estimates was performed using METAL. The methylGSA R package was used for gene set enrichment analysis. Confirmation of genome-wide significant CpG sites was performed in a cohort of Indian Asians (LOLIPOP, UK).
Results
The meta-analysis identified 76 CpG sites that were differentially methylated in individuals with incident type 2 diabetes compared with control individuals (p values <1.1 × 10−7). Sixty-four out of 76 (84.2%) CpG sites were confirmed by directionally consistent effects and p values <0.05 in an independent cohort of Indian Asians. However, on adjustment for baseline BMI only four CpG sites remained genome-wide significant, and addition of the 76 CpG methylation risk score to a prediction model including established predictors of type 2 diabetes (age, sex, BMI and HbA1c) showed no improvement (AUC 0.757 vs 0.753). Gene set enrichment analysis of the full epigenome-wide results clearly showed enrichment of processes linked to insulin signalling, lipid homeostasis and inflammation.
Conclusions/interpretation
By combining results from five European cohorts, and thus significantly increasing study sample size, we identified 76 CpG sites associated with incident type 2 diabetes. Replication of 64 CpGs in an independent cohort of Indian Asians suggests that the association between DNA methylation levels and incident type 2 diabetes is robust and independent of ethnicity. Our data also indicate that BMI partly explains the association between DNA methylation and incident type 2 diabetes. Further studies are required to elucidate the underlying biological mechanisms and to determine potential causal roles of the differentially methylated CpG sites in type 2 diabetes development.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.

Introduction
Type 2 diabetes is a complex metabolic disease characterised by chronically elevated blood glucose levels, insulin resistance and beta cell failure and their interaction with obesity and physical inactivity [1,2,3]. Recent genome-wide association studies identified over 400 genetic variants associated with type 2 diabetes; however, these variants explain only a minor part of the type 2 diabetes heritability [4]. To identify missing components of type 2 diabetes aetiology, researchers started to examine gene–environment interactions and epigenetic mechanisms [5,6,7]. Improving the prediction of incident type 2 diabetes using epigenetic markers could help tailor prevention efforts focused on those at the highest risk. Moreover, epigenetics could also elucidate new pathophysiological pathways involved in type 2 diabetes development.
Recent epigenome-wide association studies (EWASs) in blood have identified differentially methylated CpG sites (DMS), in individuals with vs without type 2 diabetes, in genes such as TXNIP, ABCG1 and SREBF1 [8,9,10]. Further replication in a case–control sample of an independent cohort study confirmed the robustness of those associations with type 2 diabetes [11]. However, most of the EWASs reported so far used a cross-sectional approach, whereas it is well-known that type 2 diabetes develops over a timespan of many years before it is clinically manifest [1]. At present, only two studies examining methylation changes prior to type 2 diabetes onset have been reported: the first in the LOLIPOP cohort including 2664 participants [8]; and the second in EPIC-Norfolk including 1264 participants [12]. In both studies, increased methylation in the ABCG1 and SREBF1 genes and decreased methylation in the TXNIP gene at baseline were associated with incident type 2 diabetes.
The aim of this study was to identify additional DNA methylation markers for incident type 2 diabetes. For this, we combined results from five European prospective cohorts to increase statistical power with a focus on European ancestry in the discovery stage. The cohorts involved are the Doetinchem Cohort Study [13] from the Netherlands, the ESTHER (Epidemiologische Studie zu Chancen der Verhütung, Früherkennung und optimierten Therapie chronischer Erkrankungen in der älteren Bevölkerung) [14] and KORA (Cooperative Health Research in the Region Augsburg) [15] cohort studies from Germany and the EPIC (European Prospective Investigation into Cancer) Norfolk [16] study from the UK. We conducted a meta-analysis using DNA methylation data from EWASs obtained from blood samples collected 7–10 years prior to type 2 diabetes diagnosis. A total of 1250 cases and 1950 controls were included in this meta-analysis. Furthermore, the significant DMS obtained from the meta-analysis were tested for replication in a longitudinal cohort of Indian Asians (The London Life Sciences Prospective Population Study [LOLIPOP]) to evaluate the robustness of the associations observed [8].
Methods
Participating cohorts
In the EWAS meta-analysis we included five European cohorts (one from the Netherlands, three from Germany and one from the UK). The cohorts involved were the Doetinchem Cohort Study [13], ESTHER [14], KORA [15] and EPIC-Norfolk [16]. Two independent subcohorts from the KORA cohort were selected for EWAS analyses, designated as KORA1 (including KORA F4 and FF4 studies) and KORA2 (including KORA S3 and S4 studies). In total, five independent EWASs for incident type 2 diabetes were performed. Replication was performed in a cohort study of Indian Asians (LOLIPOP) from London, UK [8]. A general description of the cohort and characteristics of the individuals included in the current study are presented in Tables 1 and 2 (see electronic supplementary material [ESM] Methods for further details). All participants provided informed consent and the studies were approved by ethics committees.
Type 2 diabetes diagnosis
The EWAS in Doetinchem, ESTHER, KORA1, KORA2 and LOLIPOP were performed as nested case–control studies of incident type 2 diabetes, with controls matched on age, sex and measurement round. In EPIC-Norfolk, EWAS was performed as a nested case-cohort study with random selection of non-cases. In all cohorts, participants with prevalent type 2 diabetes at baseline were excluded (Table 1). Definitions of incident type 2 diabetes cases and controls varied between cohorts (Table 1). Further details are listed in Table 1 and ESM Methods (Phenotype and covariates).
Methylation measurements and quality control
DNA extracted from whole blood was bisulphite converted and hybridised to Illumina Infinium Methylation arrays (either the 450K array [KORA, ESTHER, EPIC-Norfolk, LOLIPOP] or the EPIC array [Doetinchem]). Quality control and normalisation of methylation data was conducted by each cohort separately using their own pipeline; details for each cohort are given in ESM Methods.
Cohort-specific statistical analysis
For each cohort, we independently ran EWAS models according to the same standardised analysis plan (ESM Methods), using robust linear regression models. Normalised β values for methylation intensity at each individual CpG site were modelled as the dependent variable and incident type 2 diabetes as a binary predictor variable. Additional covariates included age, sex, estimated cell types using the Houseman algorithm [17] and batches (model 1). Additionally, we adjusted the model for baseline BMI (model 2). In sensitivity analyses, both model 1 (model 1.1) and model 2 (model 2.1) were additionally adjusted for smoking (three categories: current; never; ever smoker) and follow-up time (years between sample collection for DNA methylation measurements and diagnosis of type 2 diabetes [equivalent year for matched controls]). For additional models we calculated percentile reduction/attenuation of effect sizes compared with model 1.
Meta-analysis and replication
Inverse-variance fixed-effects meta-analyses of cohort-level individual CpG EWAS estimates were performed using METAL [18]. We corrected for multiple testing by applying a stringent genome-wide significant p value <1.1 × 10−7 (i.e. 0.05/450k). Potential heterogeneity between studies was quantified using the I2 measure (the percentage of variance explained by study heterogeneity) and CpG sites with I2 > 60% and heterogeneity p value <0.05 were highlighted. We also highlighted all significant DMS listed as polymorphic or cross-hybridising CpG sites [19]. For polymorphic CpG sites, we used Hartigan’s dip test to evaluate the possible binomial distribution of DNA methylation levels in methylation data of the Doetinchem cohort [20]. We used the HumanMethylation450 v1.2 Manifest File (https://support.illumina.com/downloads/infinium_humanmethylation450_product_files.html) and the R package ‘FDb.InfiniumMethylation.hg19’ version 2.2.0 (https://bioconductor.org/packages/FDb.InfiniumMethylation.hg19/) to annotate to the nearest gene for each CpG. Furthermore, we checked for overlap between our significant DMS and previously published EWAS results related to blood-based incident and prevalent type 2 diabetes, blood lipids, BMI and BP [8, 11, 12, 21,22,23,24,25,26,27,28,29,30]. All genome-wide significant CpG sites associated with incident type 2 diabetes were used for replication in an independent cohort of Indian Asians (LOLIPOP). CpGs were considered replicated if they had directionally consistent effects and a p value <0.05 (nominal significance). Furthermore, we checked the correlation of effect sizes between discovery and replication stages. To test the predictive ability of the 76 markers for incident type 2 diabetes as an outcome, a methylation risk score (MRS) was calculated based on the summation of the 76 CpGs weighted by the effect sizes from an alternative model of the EPIC-Norfolk dataset [12], which used incident type 2 diabetes as the dependent variable (β values represented the OR per 1% methylation change). Then, receiver operating characteristic curve analyses were performed to provide estimates for AUC in the independent LOLIPOP cohort. We tested models predicting incident type 2 diabetes by the MRS only (model M1), by established phenotypic risk factors only, including age, sex, BMI and HbA1c (model M2) and combining both (model M3). We additionally adjusted models M1, M2 and M3 for cell type distributions (models M4, M5, M6, respectively). To investigate the predictive capacity of CpG sites not reaching genome-wide significance (i.e. p>1 × 10−7), we compared AUC values from MRSs based on four increasingly lenient p value thresholds (p<1 × 10−7, p<1 × 10−6, p<1 × 10−5 and p<1 × 10−4) with increasing numbers of CpG sites. We performed those analyses in the European-ancestry Doetinchem cohort based on results from leave-one-cohort-out EWAS meta-analysis (see ESM Methods for details).
Gene set enrichment analysis, transcription factor analysis and association with gene expression
Using the full genome-wide results of model 1 from the meta-analysis, we performed gene set enrichment analysis with the methylGSA R package to relate CpG sites to their biological function [31]. We included Kyoto Encyclopedia of Genes and Genomes (KEGG) and Reactome pathways as well as Gene Ontology (GO) terms available in the package. We corrected for multiple testing using false discovery rate (FDR) <5% [32].
Next, we focused on the 76 genome-wide significant DMS and performed a transcription factor (TF) enrichment analysis using the web-based ChIP-X Enrichment Analysis 3 (ChEA3) tool [33]. The enriched TFs were ranked based on Fisher’s exact test (p value <0.01).
To additionally look-up previously reported associations of phenotypes/diseases with genetic variants located in or near associated CpG sites, we submitted a list of gene names nearest to the 76 DMS from our EWAS meta-analysis to the NHGRI-EBI GWAS Catalog (https://www.ebi.ac.uk/gwas/, accessed 25 May 2020). Similarly, we queried the list of 76 DMS in the EWAS catalog (http://www.ewascatalog.org/, accessed 15 February 2021). We highlighted associations related to metabolic traits, lipid traits, BP and obesity.
Furthermore, we investigated the association between our 76 genome-wide significant DMS, gene expression levels in blood and SNPs using publicly available expression quantitative trait methylation (eQTM) results from the BIOS consortium (https://www.genenetwork.nl/biosqtlbrowser/, accessed 9 July 2020) and methylation quantitative trait loci (meQTL) from GoDMC (http://mqtldb.godmc.org.uk/, accessed 20 July 2021).
Results
Characteristics of the meta-analysis cohorts
Baseline characteristics of the cohorts participating in the discovery meta-analysis and replication are presented in Table 2. The mean age at baseline ranged from 50.3 to 62.7 years across cohorts, and the proportion of men ranged from 42% to 68.1% for both incident type 2 diabetes cases and controls. The mean follow-up time between DNA methylation measurements in blood and type 2 diabetes diagnosis ranged from 6.25 to 10.5 years across cohorts. Already at baseline, we observed a higher mean BMI in incident type 2 diabetes cases compared with controls in all cohorts. Similarly, baseline indicators of hyperglycaemia (i.e. fasting glucose and/or HbA1c) were higher in incident type 2 diabetes cases compared with controls in ESTHER, KORA1, EPIC-Norfolk and LOLIPOP. We observed differences in smoking status between incident type 2 diabetes cases across cohorts, with the proportion of current smokers ranging from 9.4% in LOLIPOP to 36.4% in the Doetinchem cohort (Table 2).
Meta-analysis results of discovery
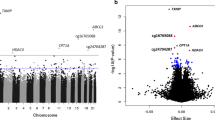
Combining the results of the five discovery EWAS, we identified 76 genome-wide significant DMS using model 1 (λ = 1.189; QQ plots per cohort and for the whole meta-analysis for all models are presented in ESM Fig. 1). Of these, 63 DMS have not been previously reported to be associated with incident type 2 diabetes. The 76 DMS were annotated to 65 genes. Some of these genes had multiple CpG sites annotated to them: LGALS3BP (5); ABCG1 (3); SYNGR1 (3); SLC9A1 (2); PFKFB3 (2); and NAP1L4 (2) (Table 3). The results are summarised in a Manhattan plot (Fig. 1), showing the distribution of CpG sites across the genome. Based on principal component analysis (PCA) performed in the Doetinchem dataset, 32 out of the 76 CpG sites were considered independent signals (90% of variance explained). CpG site cg11800635 was listed as a probe with potential cross-hybridisation and 11 CpG sites were listed as polymorphic CpGs (Table 3). However, for eight out of those 11 CpG sites available in the Doetinchem dataset, we found no evidence of binomial methylation distributions, suggesting lack of confounding by the underlying SNP (dip-test p values 0.5–0.99). Of the 76 DMS identified, 20 DMS (26%) showed I2 > 60% suggesting considerable heterogeneity between studies (p<0.05; Table 3); for each of these 20 CpG sites, we made forest plots (ESM Fig. 2). Despite high, statistically significant heterogeneity estimates, only one site showed a difference in the direction of the association between cohorts (cg19169154 in KORA1; I2 = 66.2%). Also, KORA1 showed large differences in effect size for cg19693031 (I2 = 89.2%) and cg11269166 (I2 = 79.7%). For some sites, two clusters of cohorts with similar effect sizes seemed to be present (e.g. cg24678869 [I2 = 71.4%]). Otherwise, despite the high heterogeneity estimates, effect estimates were broadly consistent between cohorts.

Manhattan plot showing 76 genome-wide significant CpG sites (above red line, p<1.1×10−7) associated with incident type 2 diabetes in five European cohorts (N=1250 cases/1950 controls). Gene annotations for the ten most significant CpG sites are indicated in the plot; y-axis shows negative log of associated p value
As a sensitivity analysis, we evaluated the impact of smoking and follow-up time from sample collection until type 2 diabetes diagnosis. With this additional adjustment (model 1.1) there was a reduction in the number of significant DMS from 76 to 47 (ESM Table 1; follow-up time not available for EPIC-Norfolk non-cases and LOLIPOP). Adjustment for baseline BMI (model 2) and for BMI, smoking and follow-up time (model 2.1) revealed that the number of significant DMS associated with incident type 2 diabetes decreased from 76 to 4 and 3, respectively (still including the two top CpG sites at the TXNIP and ABCG1 genes; ESM Tables 2 and 3). The attenuation of effect sizes across all models per CpG site is presented in ESM Table 4. Mean attenuation for all 76 CpG sites was 3% in model 1.1, while in models 2 and 2.1 the mean attenuation of effects was 22% and 26%, respectively. The correlation of effect sizes between models for all 76 DMS was very high and varied between 0.98 and 0.99 (ESM Fig. 3).
Comparison with previous EWASs of incident and prevalent type 2 diabetes, lipids, BMI and BP
Previously, 13 of the 76 DMS had been reported to be associated with incident type 2 diabetes [8, 12] and nine with prevalent type 2 diabetes [11, 24], all with consistent directions of effect (ESM Table 5). Furthermore, 33 of the 76 DMS (43%) overlapped with BMI EWAS results [21, 27,28,29,30], with consistent direction of the effects, and 12 DMS (16%) overlapped with blood lipid EWAS results, including triacylglycerols, total cholesterol, LDL-cholesterol and HDL-cholesterol [25, 26]. Additionally, five DMS (7%) had previously been reported in EWASs on BP [22, 23] (ESM Table 5).
Replication
Out of the 76 genome-wide significant DMS, 64 (84.2%) showed significant, directionally consistent association with incident type 2 diabetes in Indian Asians in model 1 (p<0.05; ESM Table 6). Using models 1.1, 2 and 2.1, 40 out of 47 (85%), three out of four (75%) and two out of three (67%) DMS, respectively, were replicated in the LOLIPOP cohort (ESM Tables 1–3). Although we observed a substantial attenuation of effect sizes of 47% in our replication (ESM Table 4), the correlation of effect sizes between discovery and replication stages was high (r = 0.91; ESM Fig. 3). Next, we combined the effects from the discovery and replication cohorts for the 76 DMS in a meta-analysis. In model 1, 63 DMS showed genome-wide significant associations with incident type 2 diabetes (p<1.1 × 10−7), whereas in models 1.1, 2 and 2.1 the number of genome-wide significant DMS increased, respectively, from 47, 4 and 3 in discovery only to 59, 18 and 10 in discovery and replication combined (ESM Table 6). Despite the high replication rate of 84.2%, we did observe considerable heterogeneity between discovery and replication, greater than that seen between discovery cohorts alone (in model 1, 53% of DMS showed significant [p<0.05] heterogeneity in combined analysis compared with 26% in discovery cohorts only).
The MRS based on 76 CpG sites showed limited predictive ability for incident type 2 diabetes (model M1, AUC = 0.591) in the LOLIPOP cohort (ESM Fig. 4). Moreover, the addition of the MRS to a prediction model including established predictors of type 2 diabetes (age, sex, BMI and HbA1c) showed no improvement (model M2, AUC = 0.753 vs model M3, AUC = 0.757). Additional adjustment for cell type distributions in these models did not change these conclusions (models M4, M5, M6). In the Doetinchem cohort we observed a slight improvement in AUC after adding an MRS based on genome-wide significant CpG sites (model M1 [age, sex, BMI, cell types, batch], AUC = 0.735; model M2 [age, sex, BMI, cell types, batch and MRS], AUC = 0.755; ESM Fig. 5). However, adding additional CpG sites based on less-stringent p value thresholds did not improve the AUC, indicating the limited predictive capacity of CpG sites that did not achieve genome-wide significance in the current meta-analysis (ESM Fig. 6).
Gene set enrichment analysis and associations with gene expression and SNPs
The results of gene set enrichment analyses based on genome-wide DNA methylation results from model 1 are presented in ESM Tables 7–9. The insulin signalling pathway was enriched in KEGG analysis, although the association did not survive the FDR correction (FDR = 0.12). Furthermore, fatty acid and lipid homeostasis appear to be perturbed in future type 2 diabetes cases, since pathways such as phospholipid metabolism and metabolism of steroids were found to be enriched (Reactome analysis, FDR = 0.04; GO terms, FDR < 0.05). As a sensitivity analysis we repeated the gene set enrichment analyses on the fully adjusted model 2.1 (adjusted for BMI, smoking and follow-up time). As expected, similar pathways came up; however, the FDR significance level was not reached due to the higher p values of individual CpG sites from model 2 (ESM Tables 7–9).
Analysis of enrichment of TFs for the 65 annotated gene names out of 76 DMS, using the ChEA3 online tool, resulted in 48 TFs (p<0.01; ESM Table 10).
Further, we queried the list of 65 annotated gene names in the GWAS catalog to find previously reported associations of phenotypes/diseases with genetic variants at those loci. Seventeen out of 65 (26%) genes harboured genetic variant associations with at least one metabolic trait or disease, such as lipid traits, BP and obesity (Table 3; ESM Table 11).
Next, we queried the list of 76 genome-wide significant CpG sites in the EWAS catalog to find previously reported associations with phenotypes/diseases. Fifty-three out of 76 (70%) CpG sites were identified in EWAS studies of at least one metabolic trait and 24 (31.6%) CpG sites were previously reported to be associated with smoking (ESM Table 12).
We investigated whether DNA methylation levels of the 76 CpG sites were significantly associated with gene expression levels in blood. Of the 76 DMS identified, 21 CpG sites (28%) were associated with expression levels of 23 genes, including top signals at genes such as TXNIP, ABCG1, SREBF1 and CPT1A (Table 3; ESM Table 13). Additionally, we performed a look-up of known meQTL. Of the 76 DMS, DNA methylation at 59 CpG sites (78%) showed significant association with at least one SNP and, in total, 14,813 cis associations were found with 13,121 SNPs (p<5 × 10−8). Of these, 80 mQTL were identified after clumping (ESM Table 14).
Discussion
To the best of our knowledge, this is the first meta-analysis of methylation markers for incident type 2 diabetes. Previous studies have investigated the association between DNA methylation and incident type 2 diabetes in single cohorts [8, 12]. By combining DNA methylation data from five EWASs from European cohorts we successfully increased the power of the study and identified 76 DMS that were associated with incident type 2 diabetes.
Type 2 diabetes is a complex disease that exhibits metabolic changes many years prior to clinical disease onset. Using a prospective study design, we identified multiple changes in DNA methylation levels preceding the onset of type 2 diabetes. After adjustment for baseline BMI, we observed a large attenuation of significant CpG sites in the discovery phase. The EPIC-Norfolk study also investigated the effects of baseline BMI on their EWAS results and detected a similar reduction in the number of significant DMS [12]. However, a modest mean attenuation of effect sizes after BMI adjustment of 22% and the strong correlation of adjusted effect sizes with those of the primary discovery model (r = 0.983) suggested a smaller effect of BMI than might have been expected based only on the large reduction in number of genome-wide significant signals (reduction of 95%). Findings from a recent large EWAS focusing on BMI suggest that changes in DNA methylation profiles are a consequence of adiposity rather than a cause [27]. A look-up in the EWAS catalog revealed that 24 of our 76 top CpG sites were previously reported to be associated with smoking. This result is in line with the observed reduction in the number of significant DMS from 76 to 47 after adjustment for smoking (and follow-up time) and highlights the relevance of smoking, which not only impacts methylation but has also been reported as a risk factor for type 2 diabetes [34]. Our results show the importance of confounders such as smoking and BMI in the association between DNA methylation and type 2 diabetes. Although after adjustment for BMI effect sizes attenuate by about 20% and most CpGs lose genome-wide significance, attenuation is modest compared with the large reduction in the number of genome-wide significant signals, offering promise for future meta-analyses of larger size to significantly detect the DNA methylation signals predictive of incident type 2 diabetes that are independent of BMI.
Gene set enrichment and TF analyses performed to obtain better insight into biological mechanisms revealed perturbation of biological processes linked to insulin signalling, and fatty acid and lipid homeostasis. The results from our meta-analysis included CpG sites at genes that are known to be associated with type 2 diabetes, such as TXNIP, ABCG1, SREBF1 and CPT1A, showing consistency between cross-sectional and longitudinal studies and also between ethnicities [9, 10, 35]. However, these findings are accompanied by 63 CpG sites novel for incident type 2 diabetes annotated to a number of genes that, at least partly, also seem to be relevant for type 2 diabetes. Examples include OLMALINC, UFM1, LGALS3BP, TRIO and CFL2. OLMALINC (oligodendrocyte maturation-associated long intergenic non-coding RNA) is a long intervening non-coding RNA that was recently reported to function as an epigenetic regulator of lipid metabolism [36]. UFM1 (encoding ubiquitin-fold modifier 1) may play a crucial role in various cellular processes including endothelial reticulum stress-induced apoptosis of pancreatic beta cells [37]. LGALS3BP encodes a glycoprotein belonging to the family of galectins, which are presumed to be involved in regulating processes linked to the immune response and inflammation [38,39,40]. TRIO encodes a guanine exchange factor (trio rho guanine nucleotide exchange factor), which is a component of the Rho GTPase nucleotide cycle. Rho GTPases play a crucial role in metabolic homeostasis [41]. CFL2 has been reported to be involved in actin remodelling required for recruitment of vesicles containing GLUT4 upon insulin stimulation [42]. Thus, this meta-analysis resulted in the identification of additional DNA methylation markers for incident type 2 diabetes. However, we also observed that a large proportion of those CpG sites have previously been identified in BMI, lipid and BP EWASs, suggesting common or related (epi)genetic mechanisms underlying those associations.
We recognise several limitations of the study presented here. First, although all cohorts excluded prevalent cases of type 2 diabetes at baseline based on a number of criteria (Table 1), this was not cross-validated by glycaemic measures in the EPIC-Norfolk and parts of the KORA2 and Doetinchem cohorts. As such we cannot exclude that some incident cases in these cohorts may have had prediabetes or even undiagnosed type 2 diabetes at baseline. However, forest plots of the 20 CpG sites showing considerable heterogeneity between studies failed to reveal consistent differences due to specific cohorts, suggesting that the high heterogeneity was not primarily driven by these cohorts. Second, we focused on whole-blood DNA methylation, which may not fully represent methylation patterns in other more metabolically relevant tissues such as adipose tissue, liver or muscle. Next, we cannot rule out the possibility of reverse causation, where the DNA methylation changes we identified are a consequence of raised blood glucose levels and adiposity rather than a cause. Gradually rising levels of blood glucose and adiposity in the years prior to clinical diagnosis of type 2 diabetes may elicit compensatory epigenetic changes, reflecting increased levels of metabolic dysregulation. We chose to correct our meta-analysis results for multiple testing using the commonly applied Bonferroni correction; however, we acknowledge that other methods would have yielded other sets of significant CpG sites (e.g. Saffari et al’s [43] cut-off of p<3.6 × 10−8 would have decreased the number of significant CpGs from 76 to 59). Additionally, if we had corrected our replication analysis either for 76 tests (i.e. Bonferroni) or the number of independent signals identified through PCA (i.e. 32), the set of replicated CpG sites would have decreased from 64 to 39 and 46, respectively. Importantly, this meta-analysis of results from multiple cohorts increased the statistical power of associations between DNA methylation and type 2 diabetes compared with previous single-cohort studies.
Taken together, this large meta-analysis of EWASs resulted in the identification of 76 DMS associated with incident type 2 diabetes. The results from the replication analysis in a cohort of Indian Asians suggest that the association between DNA methylation levels and incident type 2 diabetes is independent of ethnicity. Our data also show that BMI partly explains the association between DNA methylation and incident type 2 diabetes. Functional analyses revealed multiple biological pathways involved in fatty acid and lipid metabolism, immune response and inflammation, which partly underlie impaired glucose metabolism. Further studies are required to evaluate the relevance to other tissues and to determine whether these DMS have a causal role in type 2 diabetes development. In addition, a more detailed analysis of their biological function is warranted. Future work could assess correlations between our poly-epigenetic predictor of incident type 2 diabetes and DNA methylation-based predictors of BMI and related traits, including waist/hip ratio and per cent body fat such as those generated by McCartney et al [44]. It would also be interesting to test whether such DNA methylation-based predictors add information in prediction models over and above available phenotypic analogues.
Data availability
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- DMS:
-
Differentially methylated CpG sites
- eQTM:
-
Expression quantitative trait methylation
- EWAS:
-
Epigenome-wide association study
- FDR:
-
False discovery rate
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- meQTL:
-
Methylation quantitative trait loci
- MRS:
-
Methylation risk score
- PCA:
-
Principal component analysis
- TF:
-
Transcription factor
References
Kahn SE, Cooper ME, Del Prato S (2014) Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet (London, England) 383(9922):1068–1083. https://doi.org/10.1016/S0140-6736(13)62154-6
Kahn SE, Hull RL, Utzschneider KM (2006) Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444(7121):840–846. https://doi.org/10.1038/nature05482
Mokdad AH, Ford ES, Bowman BA et al (2003) Prevalence of Obesity, Diabetes, and Obesity-Related Health Risk Factors, 2001. JAMA 289(1):76–79. https://doi.org/10.1001/jama.289.1.76
Mahajan A, Taliun D, Thurner M et al (2018) Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet 50(11):1505–1513. https://doi.org/10.1038/s41588-018-0241-6
McCarthy MI, Hirschhorn JN (2008) Genome-wide association studies: potential next steps on a genetic journey. Hum Mol Genet 17(R2):R156–R165. https://doi.org/10.1093/hmg/ddn289
Gilbert ER, Liu D (2012) Epigenetics: the missing link to understanding β-cell dysfunction in the pathogenesis of type 2 diabetes. Epigenetics 7(8):841–852. https://doi.org/10.4161/epi.21238
van Dijk SJ, Tellam RL, Morrison JL, Muhlhausler BS, Molloy PL (2015) Recent developments on the role of epigenetics in obesity and metabolic disease. Clin Epigenetics 7(1):66. https://doi.org/10.1186/s13148-015-0101-5
Chambers JC, Loh M, Lehne B et al (2015) Epigenome-wide association of DNA methylation markers in peripheral blood from Indian Asians and Europeans with incident type 2 diabetes: a nested case-control study. Lancet Diabetes Endocrinol 3(7):526–534. https://doi.org/10.1016/S2213-8587(15)00127-8
Florath I, Butterbach K, Heiss J et al (2016) Type 2 diabetes and leucocyte DNA methylation: an epigenome-wide association study in over 1,500 older adults. Diabetologia 59(1):130–138. https://doi.org/10.1007/s00125-015-3773-7
Soriano-Tárraga C, Jiménez-Conde J, Giralt-Steinhauer E et al (2016) Epigenome-wide association study identifies TXNIP gene associated with type 2 diabetes mellitus and sustained hyperglycemia. Hum Mol Genet 25(3):609–619. https://doi.org/10.1093/hmg/ddv493
Walaszczyk E, Luijten M, Spijkerman AMW et al (2018) DNA methylation markers associated with type 2 diabetes, fasting glucose and HbA1c levels: a systematic review and replication in a case–control sample of the Lifelines study. Diabetologia 61(2):354–368. https://doi.org/10.1007/s00125-017-4497-7
Cardona A, Day FR, Perry JRB et al (2019) Epigenome-Wide Association Study of Incident Type 2 Diabetes in a British Population: EPIC-Norfolk Study. Diabetes 68(12):2315–2326. https://doi.org/10.2337/db18-0290
Verschuren W, Blokstra A, Picavet H, Smit H (2008) Cohort Profile: The Doetinchem Cohort Study. Int J Epidemiol 37(6):1236–1241. https://doi.org/10.1093/ije/dym292
Raum E, Rothenbacher D, Löw M, Stegmaier C, Ziegler H, Brenner H (2007) Changes of cardiovascular risk factors and their implications in subsequent birth cohorts of older adults in Germany: a life course approach. Eur J Cardiovasc Prev Rehabil 14(6):809–814. https://doi.org/10.1097/HJR.0b013e3282eeb308
Holle R, Happich M, Löwel H, Wichmann H, MONICA/KORA Study Group (2005) KORA - A Research Platform for Population Based Health Research. Das Gesundheitswes 67(S 01):19–25. https://doi.org/10.1055/s-2005-858235
Day N, Oakes S, Luben R et al (1999) EPIC-Norfolk: study design and characteristics of the cohort. European Prospective Investigation of Cancer. Br J Cancer 80(Suppl 1):95–103
Houseman EA, Accomando WP, Koestler DC et al (2012) DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 13:86. https://doi.org/10.1186/1471-2105-13-86
Willer CJ, Li Y, Abecasis GR (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26(17):2190–2191. https://doi.org/10.1093/bioinformatics/btq340
Pidsley R, Zotenko E, Peters TJ et al (2016) Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol 17(1):208. https://doi.org/10.1186/s13059-016-1066-1
Hartigan PM (1985) Computation of the Dip Statistic to Test for Unimodality. J R Stat Soc: Ser C: Appl Stat 34(3):320–325. https://doi.org/10.2307/2347485
Wang X, Pan Y, Zhu H et al (2018) An epigenome-wide study of obesity in African American youth and young adults: novel findings, replication in neutrophils, and relationship with gene expression. Clin Epigenetics 10(1):3. https://doi.org/10.1186/s13148-017-0435-2
Richard MA, Huan T, Ligthart S et al (2017) DNA Methylation Analysis Identifies Loci for Blood Pressure Regulation. Am J Hum Genet 101(6):888–902. https://doi.org/10.1016/j.ajhg.2017.09.028
Huang Y, Ollikainen M, Muniandy M et al (2020) Identification, Heritability, and Relation With Gene Expression of Novel DNA Methylation Loci for Blood Pressure. Hypertens (Dallas, Tex 1979) 76(1):195–205. https://doi.org/10.1161/HYPERTENSIONAHA.120.14973
Meeks KAC, Henneman P, Venema A et al (2019) Epigenome-wide association study in whole blood on type 2 diabetes among sub-Saharan African individuals: findings from the RODAM study. Int J Epidemiol 48(1):58–70. https://doi.org/10.1093/ije/dyy171
Mittelstraß K, Waldenberger M (2018) DNA methylation in human lipid metabolism and related diseases. Curr Opin Lipidol 29(2):116–124. https://doi.org/10.1097/MOL.0000000000000491
Tobi EW, Slieker RC, Luijk R et al (2018) DNA methylation as a mediator of the association between prenatal adversity and risk factors for metabolic disease in adulthood. Sci Adv 4(1):eaao4364. https://doi.org/10.1126/sciadv.aao4364
Wahl S, Drong A, Lehne B et al (2017) Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature 541(7635):81–86. https://doi.org/10.1038/nature20784
Xu K, Zhang X, Wang Z, Hu Y, Sinha R (2018) Epigenome-wide association analysis revealed that SOCS3 methylation influences the effect of cumulative stress on obesity. Biol Psychol 131:63–71. https://doi.org/10.1016/j.biopsycho.2016.11.001
Demerath EW, Guan W, Grove ML et al (2015) Epigenome-wide association study (EWAS) of BMI, BMI change and waist circumference in African American adults identifies multiple replicated loci. Hum Mol Genet 24(15):4464–4479. https://doi.org/10.1093/hmg/ddv161
Reed ZE, Suderman MJ, Relton CL, Davis OSP, Hemani G (2020) The association of DNA methylation with body mass index: distinguishing between predictors and biomarkers. Clin Epigenetics 12(1):50. https://doi.org/10.1186/s13148-020-00841-5
Ren X, Kuan PF (2018) methylGSA: a Bioconductor package and Shiny app for DNA methylation data length bias adjustment in gene set testing. Bioinformatics 35(11):1958–1959. https://doi.org/10.1093/bioinformatics/bty892
Benjamini Y, Hochberg Y (1995) Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J R Stat Soc Ser B 57(1):289–300
Keenan AB, Torre D, Lachmann A et al (2019) ChEA3: transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res 47(W1):W212–W224. https://doi.org/10.1093/nar/gkz446
Maddatu J, Anderson-Baucum E, Evans-Molina C (2017) Smoking and the risk of type 2 diabetes. Transl Res 184:101–107. https://doi.org/10.1016/j.trsl.2017.02.004
Kulkarni H, Kos MZ, Neary J et al (2015) Novel epigenetic determinants of type 2 diabetes in Mexican-American families. Hum Mol Genet 24(18):5330–5344. https://doi.org/10.1093/hmg/ddv232
Benhammou JN, Ko A, Alvarez M et al (2019) Novel Lipid Long Intervening Noncoding RNA, Oligodendrocyte Maturation-Associated Long Intergenic Noncoding RNA, Regulates the Liver Steatosis Gene Stearoyl-Coenzyme A Desaturase As an Enhancer RNA. Hepatol Commun 3(10):1356–1372. https://doi.org/10.1002/hep4.1413
Lemaire K, Moura RF, Granvik M et al (2011) Ubiquitin fold modifier 1 (UFM1) and its target UFBP1 protect pancreatic beta cells from ER stress-induced apoptosis. PLoS One 6(4):e18517. https://doi.org/10.1371/journal.pone.0018517
Hong C-S, Park M-R, Sun E-G et al (2019) Gal-3BP Negatively Regulates NF-κB Signaling by Inhibiting the Activation of TAK1. Front Immunol 10:1760. https://doi.org/10.3389/fimmu.2019.01760
Xu G, Xia Z, Deng F et al (2019) Inducible LGALS3BP/90K activates antiviral innate immune responses by targeting TRAF6 and TRAF3 complex. PLoS Pathog 15(8):e1008002. https://doi.org/10.1371/journal.ppat.1008002
Thiemann S, Baum LG (2016) Galectins and Immune Responses-Just How Do They Do Those Things They Do? Annu Rev Immunol 34:243–264. https://doi.org/10.1146/annurev-immunol-041015-055402
Møller LLV, Klip A, Sylow L (2019) Rho GTPases—Emerging Regulators of Glucose Homeostasis and Metabolic Health. Cells 8(5):434. https://doi.org/10.3390/cells8050434
Hansson B, Morén B, Fryklund C et al (2019) Adipose cell size changes are associated with a drastic actin remodeling. Sci Rep 9(1):1–14. https://doi.org/10.1038/s41598-019-49418-0
Saffari A, Silver MJ, Zavattari P et al (2018) Estimation of a significance threshold for epigenome-wide association studies. Genet Epidemiol 42(1):20–33. https://doi.org/10.1002/gepi.22086
McCartney DL, Hillary RF, Stevenson AJ et al (2018) Epigenetic prediction of complex traits and death. Genome Biol 19(1):136. https://doi.org/10.1186/s13059-018-1514-1
Acknowledgements
We thank the participants of the Doetinchem Cohort Study, as well as the field workers of the Municipal Health Services in Doetinchem (C. te Boekhorst, I. Hengeveld, L. de Klerk, I. Thus, and C. de Rover) for their contribution to the data collection of this study. We also thank M. Stijnenbosch and H. Hodemaekers-Goossens for isolating and measuring the DNA samples, and we are grateful to H. S. J. Picavet who has coordinated the fieldwork since 2007, P. Vissink for logistic management and A. Blokstra for data management (all from the National Institute for Public Health and the Environment). The authors are grateful to all ESTHER participants and gratefully acknowledge the microarray unit of the Genomics and Proteomics Core Facility of the German Cancer Research Center (DKFZ) for providing the Illumina Human Methylation arrays and related services. The authors are grateful to all KORA participants and thank all members of the KORA study team. The authors are grateful to all of the participants and staff of the EPIC-Norfolk cohort. We thank the participants and research staff who made the LOLIPOP study possible.
Authors’ relationships and activities
The authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Funding
The Doetinchem Cohort Study was supported by the Ministry of Health, Welfare and Sport of the Netherlands, the National Institute for Public Health and the Environment (grant no. S/132005) and by Biobanking and Biomolecular Resources Research Infrastructure-NL (grant no. CP2011-27). The ESTHER study was supported by the Baden-Württemberg State Ministry of Science, Research and Arts (Stuttgart, Germany), the Federal Ministry of Education and Research (Berlin, Germany) and the Federal Ministry of Family Affairs, Senior Citizens, Women and Youth (Berlin, Germany). The KORA study was initiated and financed by the Helmholtz Zentrum München – German Research Center for Environmental Health, which is funded by the German Federal Ministry of Education and Research (BMBF) and by the State of Bavaria. Furthermore, KORA research was financed by a grant from the BMBF to the German Center for Diabetes Research (DZD) and a grant from the Ministry of Culture and Science of the State North Rhine-Westphalia (Düsseldorf, Germany). The diabetes part of the KORA study was partly funded by a grant from Deutsche Forschungsgemeinschaft to WR (RA 459/3-1). It was also supported within the Munich Center of Health Sciences (MC-Health), Ludwig-Maximilians-Universität, as part of LMUinnovativ. EPIC-Norfolk is supported by programme grants from the Medical Research Council (MRC) (G9502233) and Cancer Research UK (C864/A8257). The methylation data in EPIC-Norfolk was supported through the MRC Cambridge Initiative in Metabolomic Science (MR/l00002/1). EPIC-Norfolk is also supported by MRC programme awards (MC_UU_12015/1 and MC_UU_12015/2). The LOLIPOP study is supported by the National Institute for Health Research (NIHR) Comprehensive Biomedical Research Centre Imperial College Healthcare NHS Trust, the NIHR Official Development Assistance (ODA, award 16/136/68), the European Union FP7 (EpiMigrant, 279143) and H2020 programmes (iHealth-T2D, 643774). The views expressed are those of the author(s) and not necessarily those of the Imperial College Healthcare NHS Trust, the NHS, the NIHR or the Department of Health. JC is supported by the Singapore Ministry of Health’s National Medical Research Council under its Singapore Translational Research Investigator (STaR) Award (NMRC/STaR/0028/2017).
Author information
Authors and Affiliations
Contributions
EF, AMWS, MLu and HS conceived the research question. EF conducted the analysis, wrote the initial draft of the paper and incorporated co-author comments. AMWS, MLu, JvV-O and HS provided critical feedback on the analysis and revised the manuscript for important intellectual content. JCC, KKO, HG and HB contributed the data and reviewed the manuscript. VWB, YZ, JKr, SB, FD, LZ, PW and MLo conducted the analysis. YZ, SB, FD, LZ, PW, METD, XG, CG, JKo, JKr, HS P, WR, BS, MLo, WMMV and NJW provided critical feedback on the results and reviewed the manuscript. All co-authors approved the final version of the paper. MLu and HS are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Corresponding authors
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(PDF 4171 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fraszczyk, E., Spijkerman, A.M.W., Zhang, Y. et al. Epigenome-wide association study of incident type 2 diabetes: a meta-analysis of five prospective European cohorts. Diabetologia 65, 763–776 (2022). https://doi.org/10.1007/s00125-022-05652-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-022-05652-2